Gain insights into membrane protein complexes using Molecular Dynamics simulations in Flare™
Running Molecular Dynamics (MD) calculations with membrane proteins is one of the new features recently introduced within Flare. Not only do membrane proteins play a key role in a cell’s biology, but around 20% of the human genome code for them and with regards to therapeutic targets they are of significant interest.[1] Classes of membrane proteins include GPCRs and Ion Channels.
MD simulations of membrane proteins allow for the characterization and identification of interactions between the protein and the lipid bilayer, as well as giving insight into their functions while present in their native environment.[2] Having a membrane present in a simulation containing a membrane-bound protein can be beneficial for reproducing the experimental protein’s environment which can positively impact some of the protein’s tertiary structures characteristics, such as stability.
Why run Molecular Dynamics simulations in Flare?
Flare removes the technical barriers of command-line interfaces and allows users to set up molecular systems using intuitive dialogs and the 3D viewer. Users can monitor their system throughout the set-up process using the 3D viewer and modify parameters using simple textboxes and dropdown menus.
Flare also collates several PDB sources, allowing downloading and preparation of the desired protein structures within the application. MD trajectories are saved as a widely used file format and can be viewed and analyzed with ease within the application. In this way, Flare helps you keep all aspects of your MD experiment in one easy to navigate place.
Setting up Molecular Dynamics simulations in Flare
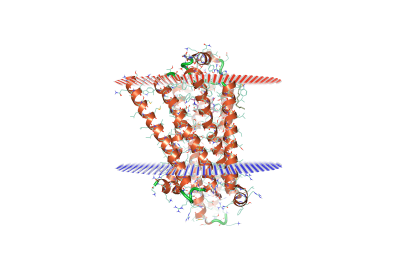
Setting up and initiating MD simulations is easy and intuitive thanks to the user-friendly GUI. Begin by downloading your PDB file from the OPM (Orientation of Membrane Proteins) [3] server, which will give an indication to where the upper and lower membrane leaflets will sit with regards to the protein.
.png)
Figure 1. Avian β1-adrenoceptor (PDB: 5a8e) with OPM membrane leaflet indications.
Once your protein has been downloaded, prepared, and undergone any manual modifications, Dynamics can be selected from the Protein tab in the GUI.
.png)
.png)
Figure 2. (Top) Example dialog box for MD calculations in Flare. (Bottom) Protein with the membrane leaflet indication in the form of a blue-outlined box.
Figure 2 (left) shows the initial dialogue box for running an MD simulation in Flare. From this, the user is able to select which protein, ligand, chain, and solvent model they’d like to run an MD simulation on, as well as the simulation length in nanoseconds. Advanced options can be accessed by clicking ‘Show Options’ at the bottom left of the dialogue box. The advanced options allow the user to modify parameters such as forcefields, box dimensions and time steps. The full range of customizable parameters can be found in the Flare user manual.
After clicking ‘Start’, Flare begins ligand parameterization (allowing for the implementation of user-defined custom parameters, if using OpenFF and forcefield initialization. Cresset currently supports OpenFF and AMBER forcefields. Once these steps are complete, Flare launches the MD calculation.
Analyzing Molecular Dynamics simulations in Flare
Upon completion of the MD simulation, the trajectory .dcd file can be analyzed from within the Flare GUI. Common analysis tools such as time-series plots of RMSD, RMSF, Potential energy and specific user-defined distances between atoms/residues are readily available, as well as contacts, secondary structure and cluster analysis.


Figure 3. Examples of post calculation analysis: (top) visual inspection (bottom left) RMSD plot, (bottom middle) RMSF of a user-specified residue, (bottom right) protein-ligand contacts.
Try Molecular Dynamics in Flare on your project
Run membrane protein MD in your own research project – request an evaluation of Flare.
References:
(1) Chavent, M.; Duncan, A. L.; Sansom, M. S. P. Molecular Dynamics Simulations of Membrane Proteins and Their Interactions: From Nanoscale to Mesoscale. Current Opinion in Structural Biology. Elsevier Ltd October 1, 2016, pp 8–16. https://doi.org/10.1016/j.sbi.2016.06.007.
(2) Stansfeld, P. J.; Sansom, M. S. P. Molecular Simulation Approaches to Membrane Proteins. Structure. November 9, 2011, pp 1562–1572. https://doi.org/10.1016/j.str.2011.10.002.
(3) Lomize, M. A.; Pogozheva, I. D.; Joo, H.; Mosberg, H. I.; Lomize, A. L. OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res 2012, 40 (D1). https://doi.org/10.1093/nar/gkr703.






















