Using molecular dynamics to produce an ensemble of protein conformations for more biologically relevant docking experiments

Introductory Overview
Earlier today, our Application Scientists, Abhijit Kayal and Ryuichiro Hara, led an exclusive online webinar session, focused on demonstrating how Molecular Dynamics and docking can work in synergy as an advanced research technique. These are two popular capabilities within Flare™, Cresset’s comprehensive platform for ligand-based and structure-based drug design.
Many proteins are, by their nature, flexible, and adopt different conformations under changing physiological conditions. This flexibility is often disregarded in traditional docking simulations, making it difficult to generate reliable docking poses for the ligand series under investigation.
Bringing a new drug to the marketplace can take over a decade, costing billions of dollars. It is one of the most complex and costly endeavors in the scientific world, with a 90% failure rate in clinical trials. As indicated in Figure 1, the cost has significantly increased over recent years. However, structure-based drug design techniques can enable promising drug candidates to be identified more efficiently and effectively.
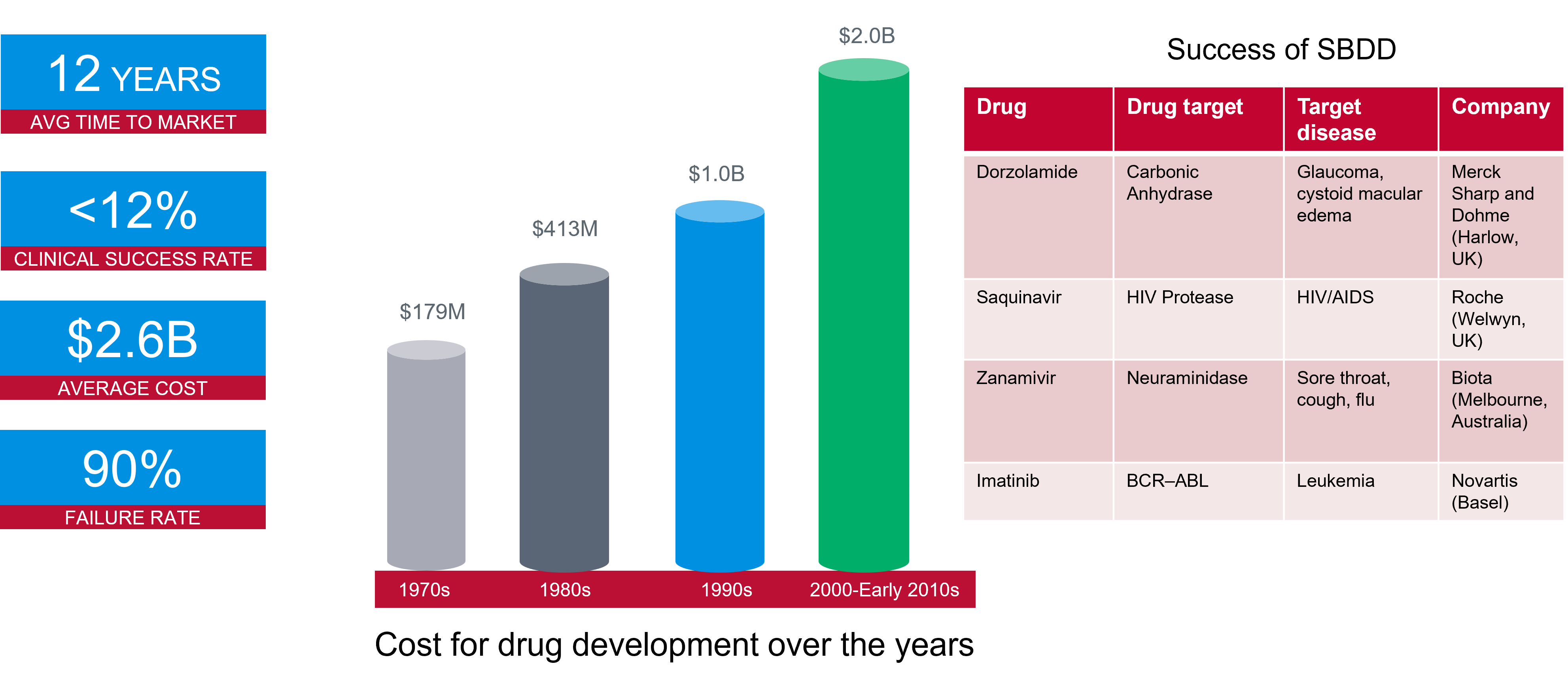
Figure 1. Success rates and costs of drug development, alongside some example structure-based drug design projects.
For those unable to attend the webinar, or looking to catch up, this article serves to summarize the session, discussing the role that Molecular Dynamics can play in generating a collection of protein conformations to which subsequent docking calculations can be applied. Continue reading to see how this technique can significantly help improve the accuracy of docking results, and ultimately accelerate the discovery of new drug molecules.
(We have annotated the headings with the timings of each section of the webinar, the recording of which can be requested, via this link.)
The challenges posed by structure-based drug design [16:40]
Abhi introduced the main webinar topic, by outlining some of the challenges posed by protein-ligand docking simulations. With a wide number of scoring methods available to evaluate the binding energy of protein-ligand complexes, each has its own unique strengths and weaknesses.
Beyond this, a major challenge for structure-based drug designers is the role played by protein flexibility. Proteins are not static structures. Depending on the 3D structure of each conformation, and the active site architecture, different ligands may bind to the active site of the protein. For example, a larger ligand may bind to the protein active site when the active site is in an enlarged position. Different binding poses or binding affinities will very often present in different protein conformations.
Computational approaches to address protein flexibility [24:14]
Several different approaches are available to help incorporate protein flexibility within docking experiments, summarized within Table 1, below.
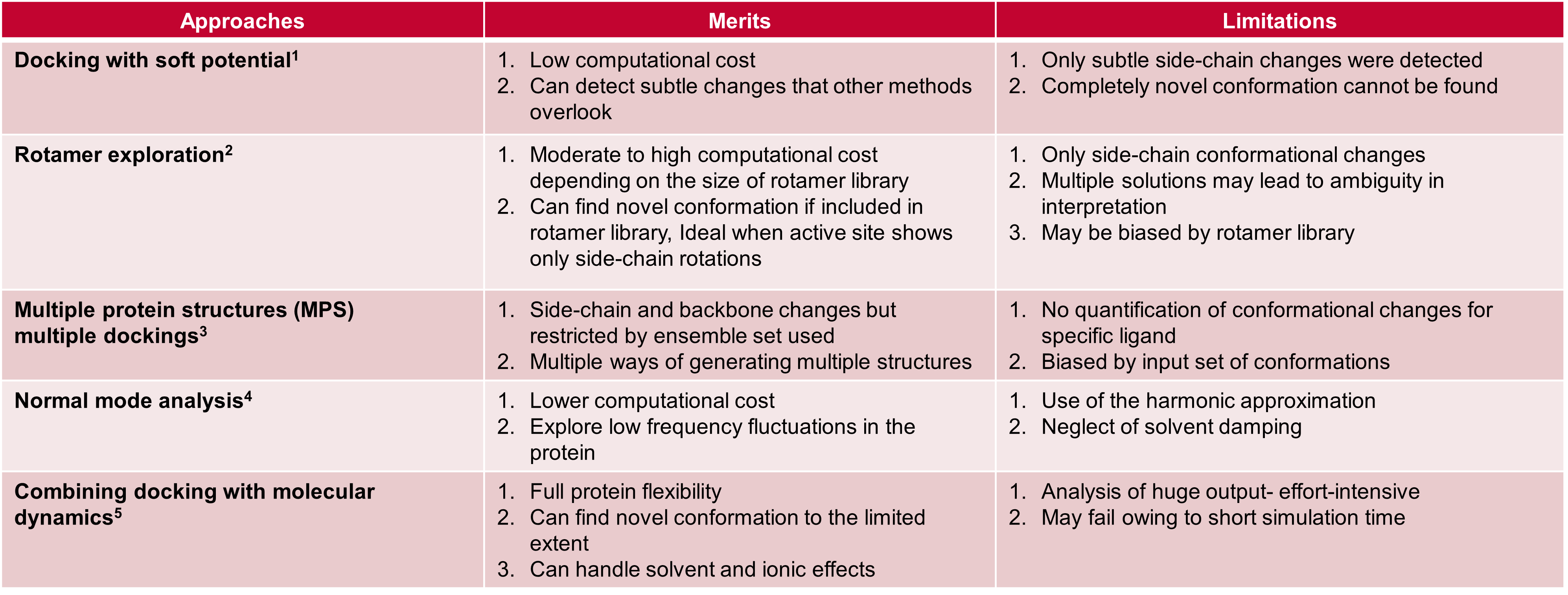
Table 1. Different approaches for incorporating protein flexibility within docking experiments.
Introduction to Cresset tools [28:26]
Abhi next took a deeper dive into the full range of features available within Flare, supporting users to prioritize the best molecules to make. Popular features within this software package include:
- QSAR modeling
- R-group analysis
- Molecular Dynamics
- Water analysis
- Pocket analysis
- Docking and scoring
- Ligand alignment
Docking and scoring in Flare uses the Lead Finder™ method, which combines a genetic algorithm search with local optimization procedures, efficiently providing promising docked poses. Three different scoring functions are used to prioritize the best poses:
- Rank score: accurate energy ranking of ligand poses
- dG: protein-ligand binding energy
- VS: rank-ordering of active/ inactives in virtual screening experiments
Ensemble docking is an advanced capability in Flare, which considers the flexibility of the protein active site within your docking experiment. Multiple conformations of the same protein can be included, and docking experiments run on each of these. The best poses across all protein conformations are saved as docked results.
Understanding the dynamic stability of protein conformations is a fundamental aspect of protein research, as it plays a key role in elucidating protein-ligand interactions and the mechanism of protein function. Flare, with its dedicated interface for creating, analyzing, and visualizing Molecular Dynamics trajectories, provides researchers with a powerful tool for studying the dynamic behavior of proteins. Based on the widely used OpenMM1 framework, Flare enables detailed investigations of protein conformational changes and their underlying energetics, thus advancing our understanding of protein dynamics and function. Flare can automatically generate custom torsion parameters for small molecules using GFN2-xTB2,3 extended semi-empirical tight-binding or ANI-2x4,5 deep learning QM approximation which significantly improves the results.
Abhi demonstrated the synergistic benefits of Flare’s ensemble docking and Molecular Dynamics capabilities, through the following two case studies.
Case studies [33:55]
The first case study focused on Cyclin-dependent kinase 2 (CDK2). In an initial self-docking experiment, two starter ligands were taken and docked against their respective crystallographic protein conformations.
- Ligand HMD, docked to structure: PDB ID: 1DM2
- Ligand STU, docked to structure: PDB ID: 1AQ1
While both protein-ligand combinations do successfully dock, cross-docking (i.e., attempting to dock ligand HMD to structure: PDB ID: 1AQ1) fails for these two ligand molecules, demonstrating that a single protein conformation is unable to predict the correct binding poses6.
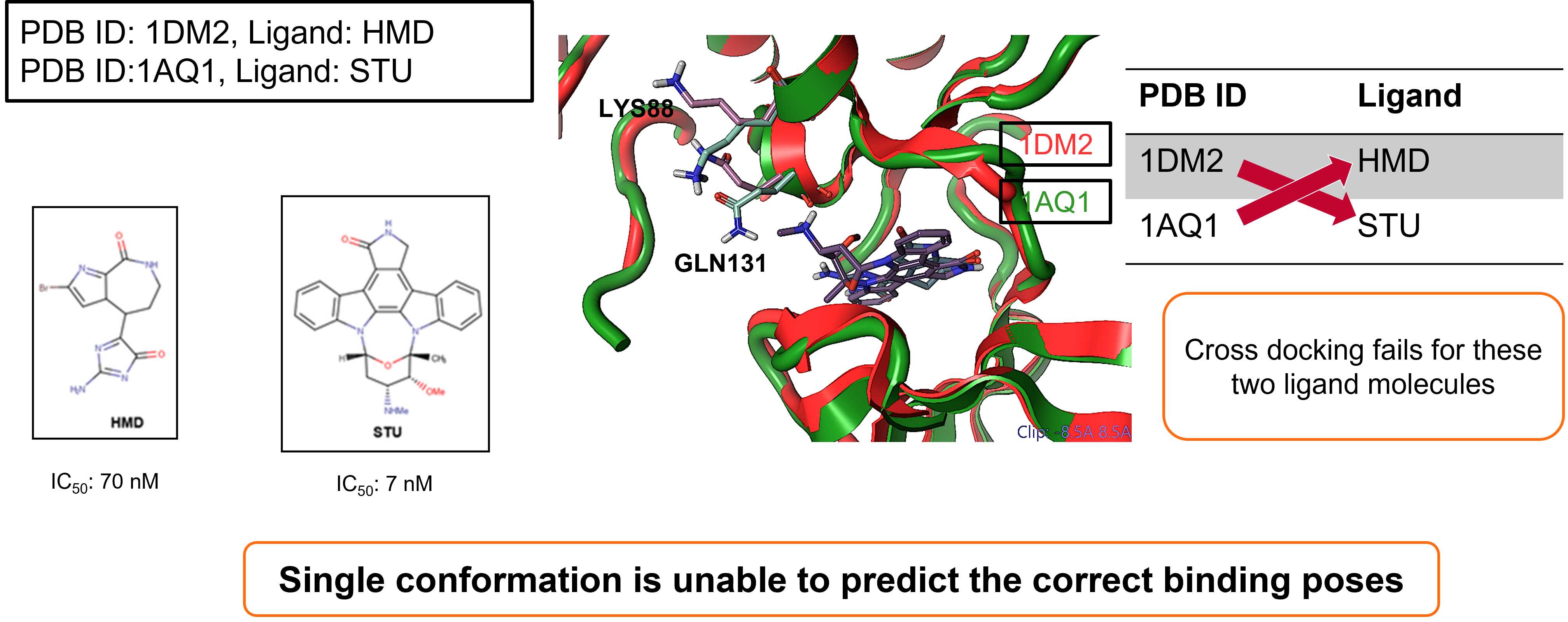
Figure 2. Cyclin-dependent kinase 2 (CDK2) docking case study.
The second case study focused on System II: Factor Xa (FXa). Once again two ligands were taken and successfully docked against their respective crystallographic protein conformation in a self-docking experiment.
- Ligand 4PP, docked to structure: PDB ID: 1XKA
- Ligand: FXV, docked to structure: PDB ID: 1KSN
Also in this case study, however, cross-docking failed for these two ligand molecules, providing further support for the need to consider multiple protein conformations within docking experiments.
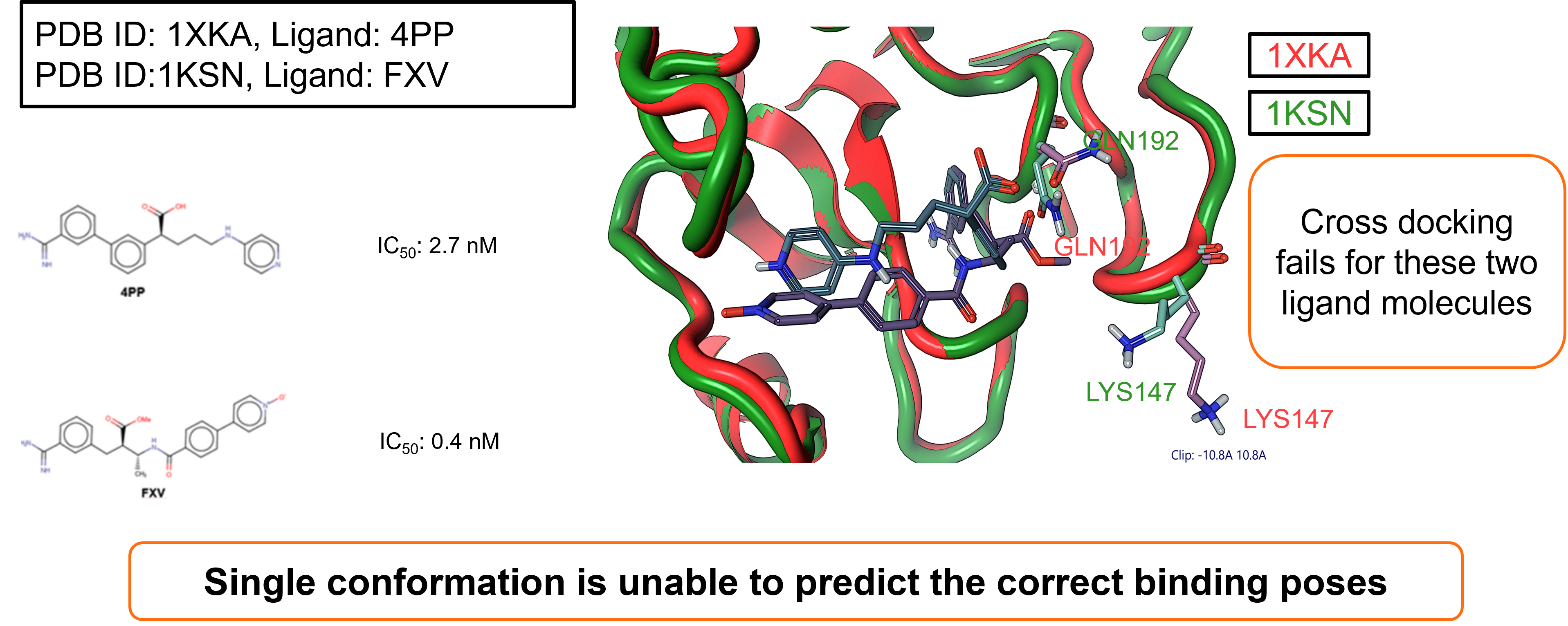
Figure 3. System II: Factor Xa (FXa) case study.
The Cresset workflow: Molecular Dynamics and ensemble docking [38:20]
To successfully consider multiple protein conformations within docking experiments, the following workflow (illustrated in Figure 4) can be applied.
- Obtain the crystal structure from a Protein Data Bank or generate a 3D structure using homology modeling.
- Prepare the protein: for example, add hydrogens/ undertake loop modelling to fully describe the structure of the protein.
- Run Molecular Dynamics simulations: to generate Molecular Dynamics trajectories which can subsequently be clustered and used to obtain multiple representative protein conformations.
- Each protein conformation can subsequently be used in the ensemble docking experiment. (Prior to the ensemble docking of each conformation, the protein atoms near the active site (within 8Å from the ligand) should be minimized.)
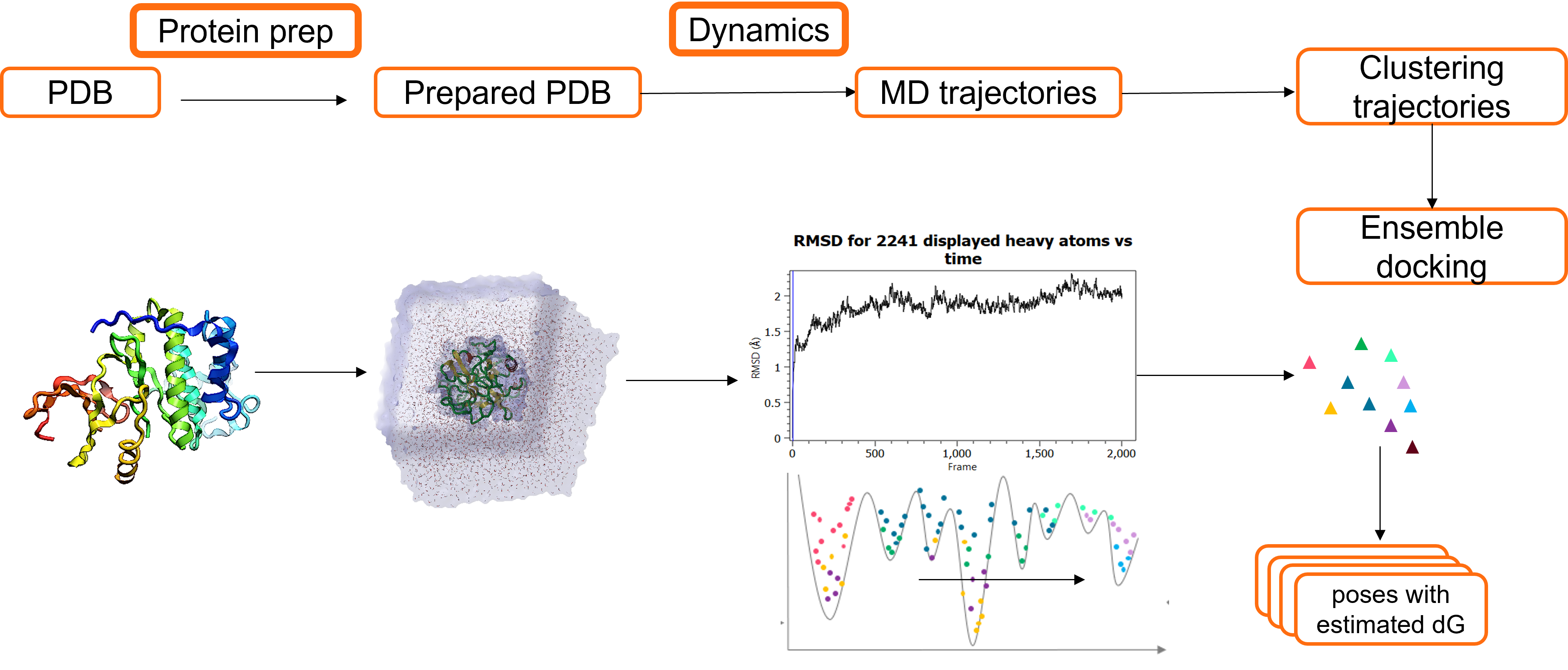
Figure 4. Molecular Dynamics and ensemble docking workflow.
Results and discussions [42:35]
The two tables in Figure 5 show the results of the self- and cross-docking experiments performed on the original X-ray structures and on the ensemble of protein conformations from Molecular Dynamics for both case studies.
Comparatively, the cross-docking ensemble docking experiments generated docked poses much closer to the crystallographic pose, with a significant improvement in estimated binding affinity.
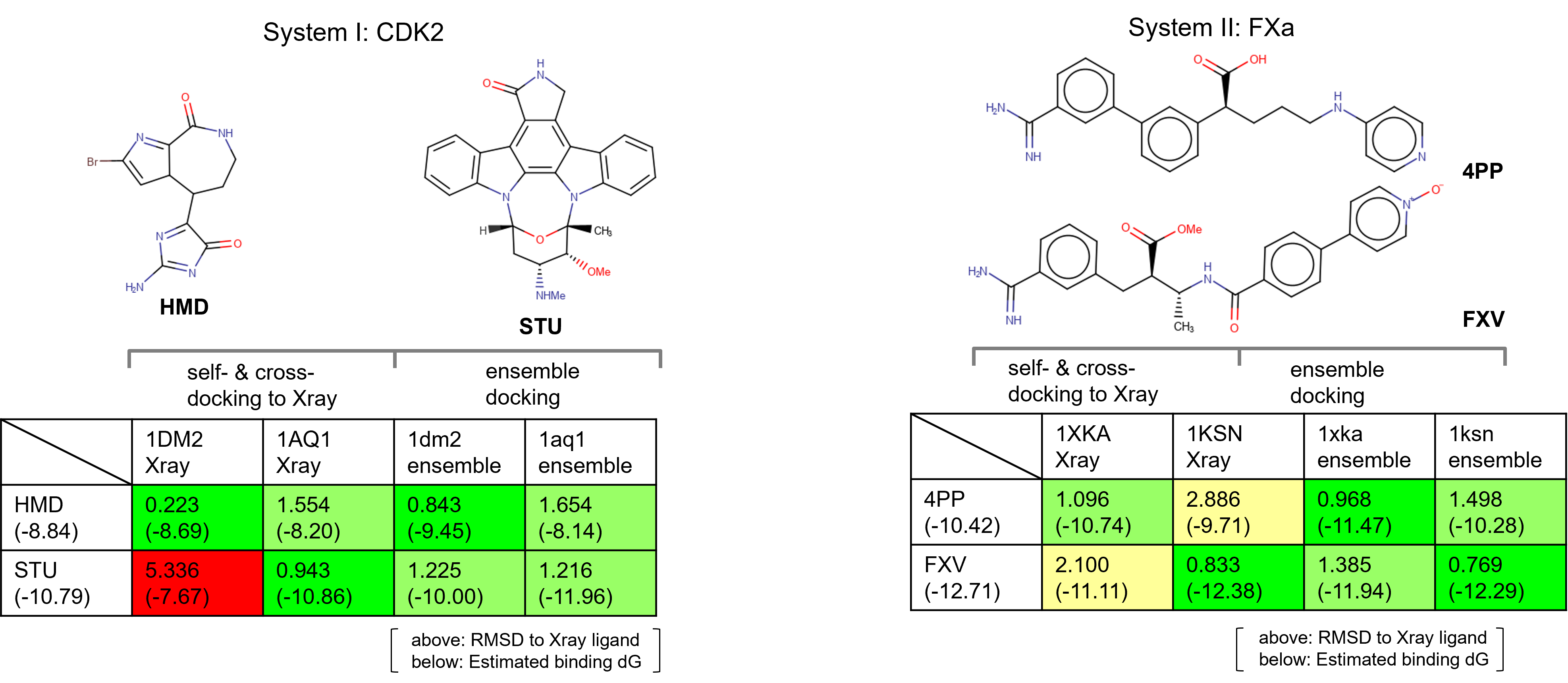
Figure 5. Ensemble docking results in comparison to self- and cross-docking to Xray.
Conclusion
Protein flexibility is inherent in nature, and most protein binding sites will contain flexible residues. It is therefore crucial to consider this flexibility when designing new ligands via in-silico methods. Flare is a powerful tool for performing ensemble docking in combination with Molecular Dynamics simulations. Request a Free evaluation of Flare today to explore these features, alongside all other available components in more depth.
The recording
Access to the full recording of the webinar discussed in this article, alongside all other recent Cresset software webinar sessions can be requested via this page.
References
- Eastman, P.; Swails, J.; Chodera, J. D.; McGibbon, R. T.; Zhao, Y.; Beauchamp, K. A.; Wang, L.-P.; Simmonett, A. C.; Harrigan, M. P.; Stern, C. D.; others. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS computational biology 2017, 13 (7), e1005659.
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended Tight-Binding Quantum Chemistry Methods. Wiley Interdisciplinary Reviews: Computational Molecular Science 2021, 11 (2), e1493.
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-XTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. Journal of chemical theory and computation 2019, 15 (3), 1652–1671.
- Smith, J. S.; Isayev, O.; Roitberg, A. E. ANI-1: An Extensible Neural Network Potential with DFT Accuracy at Force Field Computational Cost. Chemical science 2017, 8 (4), 3192–3203.
- Devereux, C.; Smith, J. S.; Huddleston, K. K.; Barros, K.; Zubatyuk, R.; Isayev, O.; Roitberg, A. E. Extending the Applicability of the ANI Deep Learning Molecular Potential to Sulfur and Halogens. Journal of Chemical Theory and Computation 2020, 16 (7), 4192–4202.
- Campbell, A. J.; Lamb, M. L.; Joseph-McCarthy, D. Ensemble-Based Docking Using Biased Molecular Dynamics. Journal of chemical information and modeling 2014, 54 (7), 2127–2138.



















