Helping to understand protein conformations with molecular dynamics

In order to get the most insight from molecular dynamics it’s important to have background understanding of the system of interest and of protein movements in general.
Finding the correct conformation: A problem of many moving parts
Proteins are highly dynamic systems. It‘s easy to forget that a single crystal structure is a static snapshot of a single conformation from an ensemble of available conformational states. In addition, there’s no guarantee that the crystallographically observed conformation is representative of the bioactive conformation rather than an artefact from the crystal growing process or due to crystal packing forces. In the same way the ligand binding mode, if present, may not be truly representative of the biologically relevant conformation.
Due to the complexity of protein and ligand movements, it would not be possible to generate all the available biologically relevant conformational states from the crystallographically observed conformational state(s). Even when there are a series of related crystal structures in differing conformational states, this still does not give the complete picture of all the available protein conformations or indeed any information on the protein energy landscape, so the energy barriers and population states of the observed conformations remain unknown. This is when molecular dynamics can help fill in the missing pieces of the conformational puzzle and protein energy landscape.
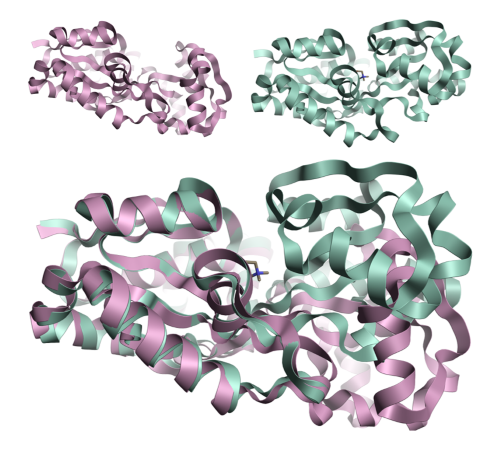
Figure 1: Crystal structures 1sw2 (top right) and 1sw5 (top left) modeled in Flare™[1] showing the protein movement observed upon ligand binding for the osmoprotection protein ProX[2]. The movement is in the relative position of the two domains rather than a restructuring of either domain.
Assessing protein stability: How can molecular dynamics help?
Molecular dynamics calculations help to map the energy landscape by solvating the protein with explicit water molecules then adding energy into the system to allow it to move, generating an ensemble of protein structures. During this process snapshots of the protein are taken at predefined time steps, calculating a series of protein structures which are likely to be representative of the biologically relevant protein conformations. Analysis of these conformations from a molecular dynamics calculation can enable the assessment of protein movements and the identification of different lower energy conformational states for the system. Because there are explicit water molecules in the system, it’s possible to identify features like bridging water molecules and those water molecules which are held rigid by the protein and, therefore, should be treated as structural features.
Like all molecular mechanics methods, the quality of the simulation depends on many factors, but two key determining factors are the quality of the force field used and the quality of the starting system, therefore, correct preparation the protein system is critical.
Currently a significant use for molecular dynamics is in the field of Free Energy Perturbation calculations (FEP). The current fashion for using pure molecular dynamics calculations is to run multiple simulations to generate a collection of energy landscapes for exploring the available conformations. This type of approach requires a large CPU or GPU farm to distribute hundreds or thousands of discrete calculations. Generating a large number of structures from a collection of discrete molecular dynamics calculations allows for more confidence in the obtained results and less susceptibility to the numerical noise that can occur in a single calculation. This approach can provide a greater understanding of the available conformations but requires extensive time, computational resource and monetary commitment. The availability of cloud computing has opened the ability to run this type of large-scale molecular dynamics calculation to a larger market, but the time and resource required to fully analyze the calculation is still extensive.
Assessing side chain mobility
Often in a series of crystal structures for a protein with a set of closely related ligands bound, the only movement observed is in those residues directly interacting or adjacent to the bound ligand. These series of crystal structures help us to assess the side-chain mobility in the active site and can assist with improving design of the current ligand series but are of less use when trying to exploit unexplored regions of the active site to develop novel series.
Systems where the active site is rigid, with only side chain movements and minimal backbone movements, can be relatively simple to investigate using a constrained molecular dynamics calculation. In these calculations only the side chains of the residues in the active site and slightly more distance co-operative residues are allowed to move. The backbone atoms and remote side chains can be either constrained or completely fixed. This allows a rapid investigation of the side chains which are known to adapt upon ligand binding. This type of calculation can also once again provide useful information on water molecules observed in or around the active site in the crystal structure, allowing for the identification of those water molecules which are stable and should be considered as structural, as opposed to those which are transient.
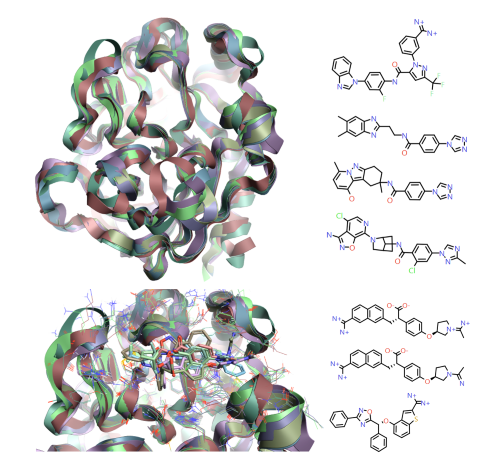
Figure 2: 1fax[3], 1mtw[4], 1x7a[5], 3lc5[6], 4yzu[7], 4z0k[7] and 5tnt[8] crystal structures for Factor IXa, modeled in Flare [1], show the overall structural conservation but the more subtle protein movement around the active site to accommodate the different ligand classes observed.
Movement in the secondary or tertiary structure
A more complex example is when there is movement in the secondary or tertiary structure of the protein and the ligand occupies a different conformational state of the protein from that available in the crystal structures. The typical example of this is in kinase systems with the DFG loop in or out conformation, but even with these reasonable well represented systems in the PDB database the crystal structures are not always available in the correct conformation states.
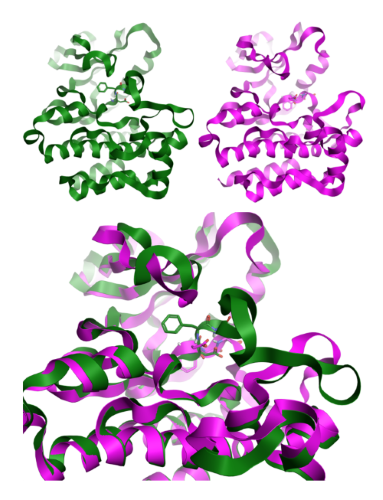
Figure 3: DFG In and Out conformations for ABL kinase, modeled in Flare [1], using crystal structures 2gqg[9] and 3qrk[10].
Working with less well represented systems
For relatively well understood and highly represented crystallographic systems like kinases it’s fairly easy to find a suitable template from which to construct a homology model of the correct conformational state. However, for less well represented systems it’s not always possible to have this wealth of structural information available.
The investigation of this type of system firstly requires an understanding of the system to appreciate the potential protein movements and conformational states available. Comparison to related family members can help to elucidate conserved protein movements and assist with this type of analysis. These identified conformational states can then be further investigated by the employment of molecular dynamics calculations to assess protein stability and the energy profile of these different conformational states and transitions between them.
Assessing the stability of homology models
Molecular dynamics is also an invaluable tool when performing homology modeling as homology models tend to be in a high energy state when first created. The regions around loop insertions and deletions and side chains generally tend to be in sub-optimum conformational states and local energy minimisation only moves these groups into the nearest energy minimum. Molecular dynamics calculations, when used properly, can be used to smooth out the energy surface and let the whole system relax from a higher energy state to a more stable lower energy state, which can be more representative of the biologically relevant system. The stability of the homology model during the molecular dynamics calculation also gives an indication of the overall quality of the homology model. Those homology models which are of lower quality tend not to survive molecular dynamics processing with their secondary or tertiary structure intact.
Contact us for a free confidential discussion
Contact Cresset Discovery Services for a free confidential discussion to see how we can help you gain greater understanding of your protein ligand systems.
References
- Flare structure-based design platform https://www.cresset-group.com/software/flare/
- Schiefner, A., Holtmann, G., Diederichs, K., Welte, W., Bremer, E. (2004) J.Biol.Chem. 279: 48270-48281
- Brandstetter, H., Kuhne, A., Bode, W., Huber, R., von der Saal, W., Wirthensohn, K., Engh, R.A (1996) J.Biol.Chem. 271: 29988-29992
- Stubbs, M.T., Huber, R., Bode, W. (1995) FEBS Lett. 375: 103-107
- Smallheer, J.M., Alexander, R.S., Wang, J., Wang, S., Nakajima, S., Rossi, K.A., Smallwood, A., Barbera, F., Burdick, D., Luettgen, J.M., Knabb, R.M., Wexler, R.R., Jadhav, P.K. (2004) Bioorg.Med.Chem.Lett. 14: 5263-5267
- Wang, S., Beck, R., Burd, A., Blench, T., Marlin, F., Ayele, T., Buxton, S., Dagostin, C., Malic, M., Joshi, R., Barry, J., Sajad, M., Cheung, C., Shaikh, S., Chahwala, S., Chander, C., Baumgartner, C., Holthoff, H.P., Murray, E., Blackney, M., Giddings, A. (2010) J.Med.Chem. 53: 1473-1482
- Parker, D.L., Walsh, S., Li, B., Kim, E., Sharipour, A., Smith, C., Chen, Y.H., Berger, R., Harper, B., Zhang, T., Park, M., Shu, M., Wu, J., Xu, J., Dewnani, S., Sherer, E.C., Hruza, A., Reichert, P., Geissler, W., Sonatore, L., Ellsworth, K., Balkovec, J., Greenlee, W., Wood, H.B. (2015) Bioorg.Med.Chem.Lett. 25: 2321-2325
- Sakurada, I., Endo, T., Hikita, K., Hirabayashi, T., Hosaka, Y., Kato, Y., Maeda, Y., Matsumoto, S., Mizuno, T., Nagasue, H., Nishimura, T., Shimada, S., Shinozaki, M., Taguchi, K., Takeuchi, K., Yokoyama, T., Hruza, A., Reichert, P., Zhang, T., Wood, H.B., Nakao, K., Furusako, S. (2017) Bioorg. Med. Chem. Lett. 27: 2622-2628
- Tokarski, J.S., Newitt, J., Chang, C.Y.J., Cheng, J.D., Wittekind, M., Kiefer, S.E., Kish, K., Lee, F.Y.F., Borzilerri, R., Lombardo, L.J., Xie, D., Zhang, Y., Klei, H.E. (2006) CANCER RES. 66: 5790-5797
- Chan, W.W., Wise, S.C., Kaufman, M.D., Ahn, Y.M., Ensinger, C.L., Haack, T., Hood, M.M., Jones, J., Lord, J.W., Lu, W.P., Miller, D., Patt, W.C., Smith, B.D., Petillo, P.A., Rutkoski, T.J., Telikepalli, H., Vogeti, L., Yao, T., Chun, L., Clark, R., Evangelista, P., Gavrilescu, L.C., Lazarides, K., Zaleskas, V.M., Stewart, L.J., Van Etten, R.A., Flynn, D.L. (2011) Cancer Cell 19: 556-568



















