Understanding the role of water in protein-ligand complexes and when to consider water in drug discovery

Introduction
Cresset Discovery has a large team of computational scientists who have the skill and experience to run detailed water analyses. By gathering valuable information from a protein structure we help you understand the role of water in a binding site and how this information can be used to rationally design ligands, explain changes in binding affinity, and in some cases adjust function e.g., from agonist to antagonist, or influence selectivity. Our research scientists can help you advance your projects efficiently and cost-effectively in the following areas:
Role and function
Understanding the role of water and its function in the binding pocket is fundamental. Water can vary from inert or can act as a reactive species and serve in biological functions. Is it bulk water, conserved water to stabilize a system, or catalytic water necessary for a specific function? For example, water molecules can mediate interactions between a protein and a substrate either to increase selectivity or lead to conformation changes. They may act as channels for proton transport or enable the hydrolysis of phosphate and sulfate substrates. These are important factors that need to be addressed to understand the water network within a binding site.
Structure and dynamics
Identification of key structural features is performed by a thorough protein preparation. A resolution of 2Å is necessary to resolve water molecules, a crystal structure with lower resolution leads to some uncertainty. Careful analysis of the electron density and/or the temperature factor might be necessary to fully assess whether water is defined (Figure 1a). Performing molecular dynamics simulations can also help identify water molecules that are truly structural from those that are transient.
Our CADD scientists will exploit structural patterns or common interactions to identify conserved water by comparing the alignment of multiple protein structures of the same protein or structures with high sequence similarities when no duplicate structure is available (Figure 1b).
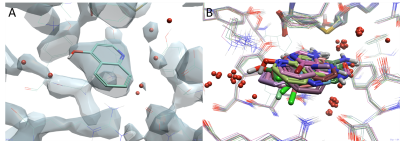
Figure 1a (left). Electron density (2Fa-Fc) for the crystal structure of hPNMT in complex with AdoHcy and 4-quinolinol (PDB code: 3KPU) with a resolution of 2.4 Å which shows some defined and undefined water molecules (red spheres). Figure 1b (right). The alignment of 14 structures of hPNMT shows the overlaid of fragment hits and clusters of conserved water molecules.
Computational methods
The position and stability of water molecules in a binding site can be calculated computationally. Cresset Discovery scientists use two water analysis methods implemented in Flare™ 1,2,3:
- GIST (Grid Inhomogeneous Solvent Theory) uses molecular dynamics simulation to analyze water stability. It can be used both on empty and liganded proteins and in each case generates heat surface maps that highlight regions of high and low water stability.
- 3D-RISM (Reference Interaction Site Model) utilizes the advanced inter-molecular descriptions of the XED force field or the AMBER GAFF force field to give a water analysis across the whole protein or can be used to place waters within the active site, with or without a ligand present.
Ligand design
Information obtained from the water analysis can be used to guide ligand design by identifying water molecules that should be ignored, or might be targeted for replacement/displacement by an additional functional group. Water molecules that are mobile and easy to replace are usually referred to as ‘hot’ or ‘unhappy’ waters, versus water that is tightly held and hard to replace referred to as ‘cold’ or ‘happy’ waters. Knowing which water molecules are energetically favorable can give valuable insights into druggable regions of the binding site and the best positions for ligands. Water in a hydrophobic environment, which is weakly polarized, will be easy to remove and tend to be the most advantageous to increase the enthalpic contribution to binding in ligand design; water that shows polar interactions with proteins residues and is strongly polarized will be hard to remove. It may also be governed by multiple factors that can compensate for each other (enthalpy‐entropy compensation), which highlights the complexity of drug design.
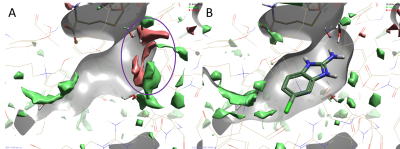
Figure 2. Results of a 3D-RISM analysis of hPNMT in complex with AdoHcy and 2-amino-5(6)-chloro-benzimidazole (PDB code: 3KR1) showing ΔG iso-surfaces map regions. Color coding: red = regions of unhappy (positive ∆G) hydration, green = regions of happy (negative ∆G) hydration. A. 3D-RISM map in the absence of the ligand shows druggable regions of the protein (red regions). B. 2-amino-5(6)-chloro-benzimidazole fragment (Kd 1.8 µM) occupies a region where water molecules are less stable and easier to displace.
As your CRO, our expert molecular modelers will collaborate with your team and apply the most appropriate approach. They will consider the flexibility of a system, the speed of the calculation plus accuracy and reliability of the method, to carefully analyze your system and assess the importance of each water molecule around a ligand. The work undertaken will help advance your project and make a significant impact on your drug design project, delivering efficiencies to ensure you only make molecules that matter.
Contact Cresset Discovery for a free confidential discussion to see how a detailed water analysis can add value to your project.
References
1. Flare V5, Cresset®, Litlington, Cambridgeshire, UK; http://www.cresset-group.com/flare/; Cheeseright T., Mackey M., Rose S., Vinter, A.; Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation J. Chem. Inf. Model. 2006, 46 (2), 665-676; Bauer M. R., Mackey M. D.; Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes J. Med. Chem. 2019, 62, 6, 3036-3050; Maximilian Kuhn, Stuart Firth-Clark, Paolo Tosco, Antonia S. J. S. Mey, Mark Mackey and Julien Michel Assessment of Binding Affinity via Alchemical Free-Energy Calculations J. Chem. Inf. Model. 2020, 60, 6, 3120–3130
2. C. Nguyen, M. K. Gilson, T. Young, Structure and Thermodynamics of Molecular Hydration via Grid Inhomogeneous Solvation Theory, ArXiv 11084876 Phys Q-Bio. 2011
3. R. Skyner, J.L McDonagh, C.R. Groom, T. van Mourik, J. B. O. Mitchell, C. R. Groom, T. Van Mourik, A Review of Methods for the Calculation of Solution Free Energies and the Modelling of Systems in Solution, Phys. Chem. Chem. Phys. 2015, 17 (9), 6174



















