Orientate your proteins on ligand superimposition using the Flare™ Python API

Orientate your proteins on ligand superimposition using the Flare™ Python API
Written by Scott Midgley
13 April 2023
The importance of protein and ligand orientation during 3D visualization
The orientation of protein structures is a simple yet essential manipulation in any 3D visualization tool for medicinal and computational chemists. In silico workflows during early-stage hit and lead identification in drug discovery often rely on the clear visual representation of interactions between a series of small, bound ligands and the corresponding active site of the target.
Ligand alignment within Flare uses Cresset’s proprietary XED force field description of molecules for fast and effective 3D ligand alignment, as well as more traditional alignment methods based on maximum common substructure. It is also possible to superimpose ligands based on a simple selection of atoms. When working with ligands in the context of proteins, it is often advantageous to apply the transformation that superimposes two (or more) bound ligands along with their associated protein structures. This gives a superimposed view of the proteins that is not achieved by aligning any of the protein sequences, but by aligning their bound ligands.
Although Flare does not have the native ability to reorientate proteins based on a superimposition of their bound ligands, the Flare Python API allows users to create custom features through access to most of Flare’s functionalities via a Python module. This has the advantage of providing a higher degree of control over the application beyond accessing the existing features through the graphical user interface (GUI). Users can create high throughput workflows or can use the functionalities to create custom tools. The latter is the focus of the present article where we demonstrate how a Flare Python API script can be used to expand the features of the base application.
We present here a simple methodology for reorienting proteins based on an atom-based ligand alignment, using a script written in the Flare Python API.
Re-orientate a protein using a ligand superimposition
As an example, a single protein was downloaded from the PDB (4QAC) and prepared in Flare. This protein is a barrel protein consisting of a 10-fold biological multimer, with individual monomers suitable for superposition in Flare. Once the protein was prepared, all chains except for the A- and B-chains were removed along with their associated ligands in the Flare Ligands Table. The B-chain was then defined as a separate protein resulting in two objects in the Flare Proteins Table: one consisting of the original A-chain, and the second protein consisting of the original B-chain. A snapshot of this is shown in Figure 1.
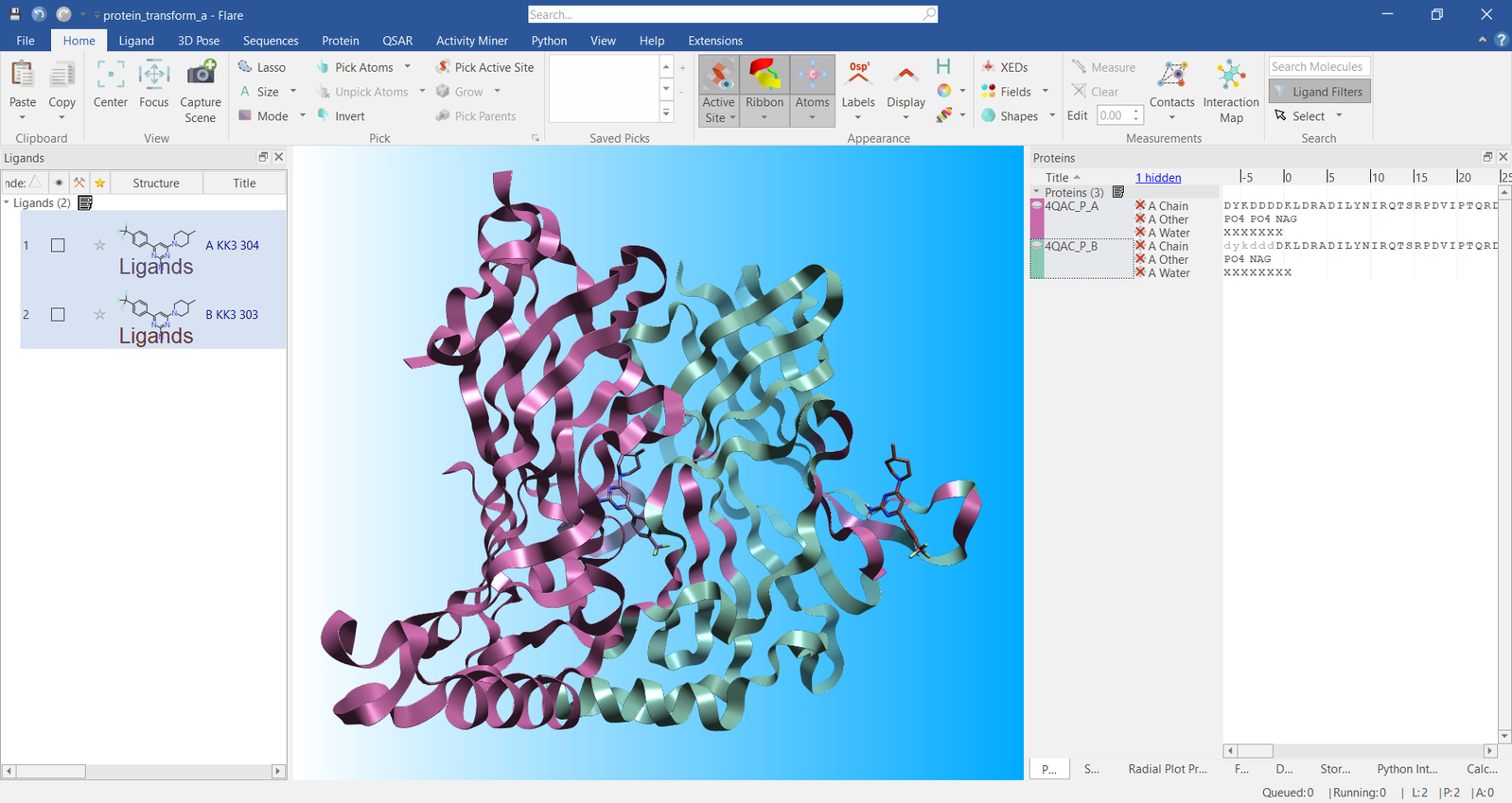
Figure 1. Visualization of the protein A- and B- chains in Flare
To begin superimposing the ligands, we first displayed the atom numbers using the keyboard shortcut, ‘Shift+i’. Figure 2 shows the two ligands with their atom numbers displayed and also confirms that they are not spatially overlapped. Flare requires at least three pairs of atoms on which to perform the ligand superimposition, but more pairs can make the overall superposition better by helping to define the 3D shape of the molecule. In this example, we used atoms 10, 8,16, and 24, as these capture longitudinal and latitudinal geometry for this ligand.

Figure 2. Spatially separated ligand molecules with atom numbers displayed.
Once we chose the atom numbers to align on, the ‘transform_qt.py’ script (which we wrote for this exercise) was loaded into the Flare Python Interpreter. To do this, we selected the ‘Python’ tab then the ‘Python Interpreter’ option, and then clicked the ‘Load…’ button in the interpreter to navigate to the script.
Before the script was run, the two ligands and two proteins were selected by clicking on the desired ligands (two) and proteins (two) while holding the ’Ctrl’ button. The script was then executed by clicking the ‘Run’ button; this provided a text box for the user to enter the atom numbers for the reference and fit molecules.
It is important to note that while the present example is being performed on the same molecules with two copies of the same protein, this script also works when superimposing two different ligands in two different proteins. Atoms with the specified numbers in the text box will be matched on vertical pairs, regardless of whether (or not) the atom numbers are the same between ligands. For example, as in Figure 3, atom 10 in the reference molecule will always be matched with atom 10 on the fit molecule, even if the ligands are different. It is therefore essential to always specify the same number of atoms in the reference box as the fit box, with a minimum of three atom numbers in each box. Each atom number should be separated by a single space.
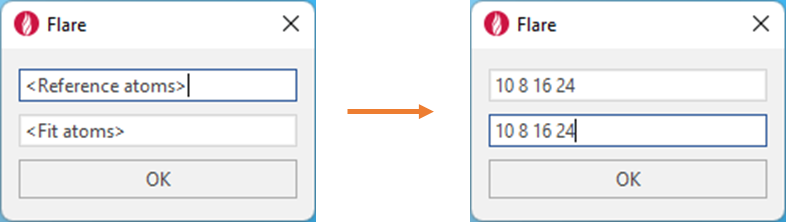
Figure 3. Defining the atom numbers to be matched from the reference molecule to the fit molecule.
The script was run by clicking the ‘OK’ button resulting in the ligands being superimposed onto the atoms specified. Immediately after the ligand superimposition, the proteins were superimposed using the same transformation matrix that superimposed the ligands. The results of this transformation are shown in Figure 4.
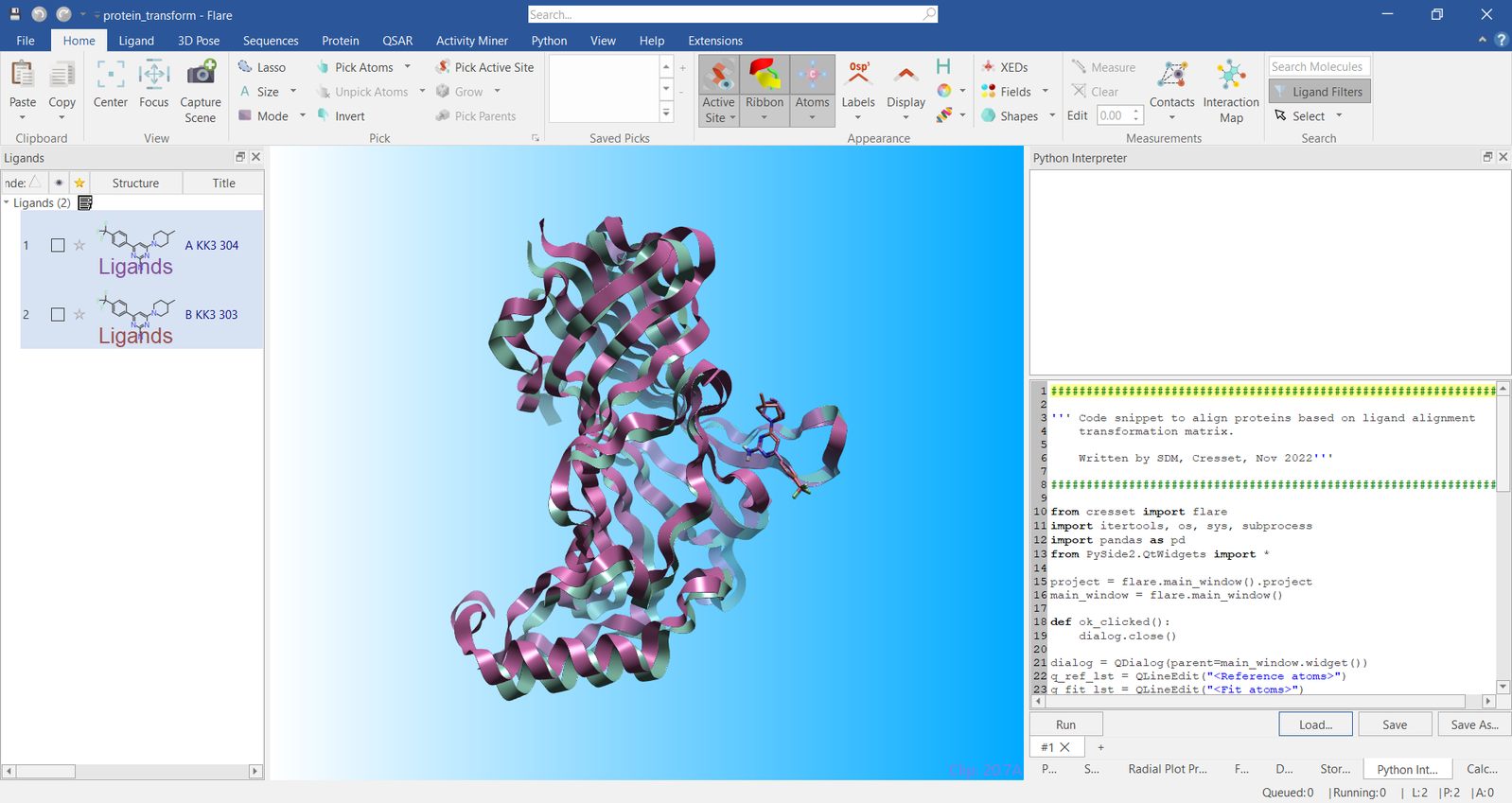
Figure 4. Visualization of the two example proteins transformed by ligand superimposition.
Summary
Flare enables the orientation of proteins to be adjusted by superimposing their bound ligands. In this article, we have demonstrated how this can be achieved for two monomers of a biological multimer. However, this workflow can be used to superimpose ligand/ligand-protein/protein pairs that are different, provided that sensible atom pairs can be identified for a ligand superimposition.
For existing Flare customers looking for more information on this functionality, or seeking to find out how to obtain the Python script used in this article, please contact Cresset support. If you’re new to Flare and would like to explore the platform’s full range of features in more depth, you can request a free evaluation.




















