Flare V5 released: Innovative science and synergy between ligand and structure-based methods

We are delighted to announce that Flare™ V5 is now available. This release of Cresset’s comprehensive platform for drug design features new and enhanced science, integration of ligand- and protein-based approaches and new and improved GUI features to increase usability.
Cutting edge QSAR methods
With this release, Flare has evolved from a structure-based solution into a comprehensive drug design platform where ligand-based and structure-based approaches work in full synergy. In Flare, you will find all your favorite qualitative and quantitative Structure Activity Relationships (SAR) methods, but better, as they benefit from protein information when available, an improved GUI and the flexibility of the Flare Python API.
The new ‘QSAR’ tab menu (Figure 1) enables easy access to all functions for building quantitative and qualitative SAR models using a choice of robust methods:
- Field QSAR using 3D descriptors modeling the shape and electrostatic character of aligned molecules, offering both prediction and interpretation thanks to informative 3D visualizations
- Machine Learning regression and classification using Support Vector Machine, Relevance Vector Machine, Random Forests, k-Nearest Neighbor, also featuring an ‘Automatic’ model building option to automatically run all the methods and pick the model with the best statistics
- Activity Atlas™ to generate qualitative 3D maps summarizing the SAR for your data series.
Relevant statistics and model information can be viewed in a dedicated ‘QSAR Model’ window.
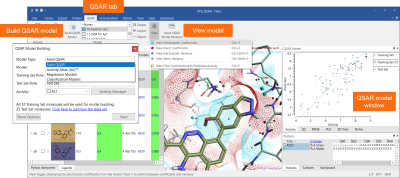
Figure 1: The QSAR tab and the QSAR Model window are a seamless interface to QSAR model building and visualization of results in Flare.
The Flare Python API makes it easy to interface Flare with an external descriptor calculator and import your favorite descriptors as additional columns into the Flare Ligands table, facilitating the development of machine learning models for predicting relevant ADMET properties.
A dedicated tab provides an enhanced interface to Activity Miner™, the Flare component enabling rapid navigation of complex SAR based on activity cliffs analysis. Multiple views of the data (Figure 2) help you find key molecule pairs in your SAR. For each pair, Activity Miner shows you how the electrostatic and shape properties differ, building an understanding of how to design better compounds with better properties.
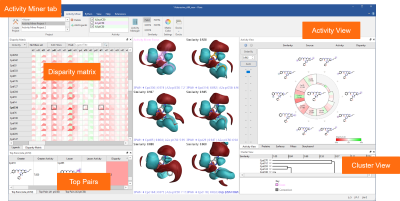
Figure 2: Activity Miner shows you multiple views of the data to help you find key molecule pairs in your SAR.
In this release, Activity Miner is fully integrated within the Flare GUI, giving access to all ligand and protein visualization choices. Multiple Activity Miner projects on multiple activity columns can be saved within same Flare project and viewed in the context of different protein structures.
Ligand-based and structure-based methods working in synergy
Ligand-based methods in Flare provide invaluable SAR information by showing you what electrostatic, hydrophobic and shape features your ligands must have to be active.
Whenever protein structures are available, viewing structure-activity information in the context of protein electrostatics gives additional insights. Protein interaction potentials, electrostatic potential (ESP) and Electrostatic Complementarity™ maps will help you see why your ligands are active, giving you an understanding into the protein’s requirements for activity which goes beyond canonical ligand-protein interactions.
Figure 3 shows two benzoxazepinone RIP1 kinase inhibitors. The oxazole ligand (left) is approximately 10 times more active than the triazole ligand (right), although the two ligands make similar ligand-protein interactions with the active site of the protein (Figure 3). Comparing the ESP map for the protein (Figure 3 - top) with that for the two ligands (Figure 3 – middle), we can see that the triazole ligand shows an area of positive electrostatics in a region where the protein is also positive. This electrostatic clash is confirmed by the Electrostatic Complementarity map for this ligand towards the RIP1 kinase active site (Figure 3 – bottom, right), and is the likely cause of the small drop in activity.
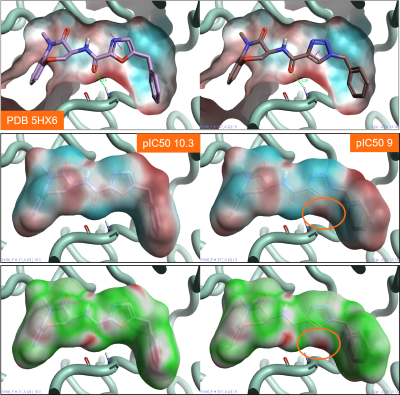
Figure 3: Top: ESP map for the RIP1 kinase active site (PDB: 5HX6). Color coding: red = positive electrostatics, blue = negative electrostatics. Middle: ESP map for oxazole (right) and triazole (left) RIP1 kinase ligands. Color coding: red = positive electrostatics, blue = negative electrostatics. Bottom: Electrostatic Complementarity maps for the same RIP1 kinase ligands. Color coding: green= good complementarity, red = electrostatic clash.
Figure 4 shows the activity cliffs summary of positive and negative electrostatics (solid surfaces) from an Activity Atlas analysis of a RIP1 kinase ligand series in the context of RIP1 kinase protein interaction potentials (wireframe surfaces). The two maps are in good agreement: the synergic application of these two methods gives us confidence that our SAR model is robust.
 from an Activity Atlas analysis of a RIP1 kinase ligand series.png)
Figure 4: Solid surfaces: Activity Cliffs Summary of ligand electrostatics for a RIP1 kinase ligand series. Wireframe surfaces: protein interaction potentials for the RIP1 kinase protein (PDB: 5HX6). Color coding: red = positive electrostatics, blue = negative electrostatics.
Water analysis with GIST
Understanding the behavior of water molecules within the active site of a protein is an extremely important aspect of drug design.
‘Druggable’ binding sites are typically occupied by water molecules which energetically would prefer to be in bulk water for both enthalpic and entropic reasons.
In a protein-ligand complex, understanding the stability of bridging water molecules is crucial to decide the drug design strategy. If the water molecule is not particularly stable, ligands may be designed to displace it and make direct interactions with the protein active site: this frequently leads to an increase in the ligand’s biological activity.
GIST is a water analysis method to assess the hydration of binding pockets and calculate the associated water thermodynamics by sampling explicit solvent distributions at the end of a molecular dynamics run. The results of a GIST analysis are shown as 3D isosurfaces mapping 'happy' (green, associated to a negative ∆G) and 'unhappy' (red, associated to a positive ∆G) regions of hydration of the active site. Unhappy regions of hydration map more druggable regions of the protein active site. Happy regions map less druggable regions where the water molecules are more stable and hence more difficult to displace.
Figure 5 – left shows the results of a GIST analysis ran on the apo active site of Acetyl-CoA Carboxylase 2 (PDB: 3GID). The results correctly map the pocket at the bottom as druggable, as expected as this region occupied is by ligand Soraphen in the PDB: 3GID protein-ligand complex. GIST also maps the binding pocket on the left side as druggable: this is confirmed by the fact that it can be occupied by alternative ligands such as ND-646 in PDB: 5KKN.
.png)
Figure 5. Results of a GIST analysis performed on the apo active site of Acetyl-CoA Carboxylase 2 (PDB: 3GID). Left: the PDB: 3GID ligand Soraphen occupies the binding pocket at the bottom. Right: the ND-646 ligand from PDB:5KKN occupies both the left and the bottom pockets. Both pockets are predicted to be druggable by GIST. Color coding: red = regions of unhappy (positive ∆G) hydration, green = regions of happy (negative ∆G) hydration.
Enhanced Flare FEP simulations to accurately predict the activity of new compounds
Flare Free Energy Perturbation (FEP) simulations in Flare V5 are significantly faster and more automated.
The faster algorithm (up to 3.5 x speedup over previous version, Figure 6) benefits from a number of system improvements which work behind the scenes:
- The implementation of an improved integrator combined with hydrogen mass repartitioning to get stable 4fs timesteps
- The simulation uses a more efficient truncated octahedral water box and a smaller solvent box buffer, making calculations faster without affecting quality of results
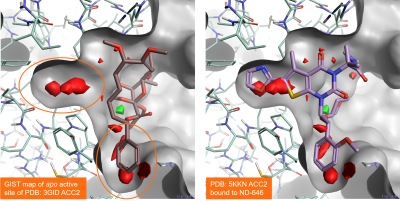
Figure 6: relative performance of Flare FEP calculations in V5 vs. Flare V4.
Flare FEP in this release is also significantly more automated. Enhancements include:
- Automatic generation of intermediate structures needed to guarantee a sufficiently smooth transformation between ligands, optimizing the success rate for the FEP simulation
- Automatic determination of the optimal lambda schedule for each transformation. Setting lambda schedules now requires minimal user interaction and provides an additional 30% overall speed improvement.
Additional efficiency is obtained by excluding from production mode calculations all ligands which do not contribute to the perturbation network.
These changes also enable Flare FEP to efficiently work at a larger scale of up to 500 ligands when working with a star graph.
Faster molecular dynamics experiments with enhanced visualization of contacts
Molecular Dynamics in Flare V5 benefits from a significantly faster algorithm, thanks to the same system improvements implemented for Flare FEP, and includes additional advanced options to fine tune the set-up of dynamics experiments:
- Improved integrator combined with hydrogen mass repartitioning to get stable 4fs timesteps
- More efficient truncated octahedral water box.
- Expanded choice of explicit water models
- Advanced options to set temperature, pressure, treatment of long-range electrostatics, non-bonded cut-off
Flare V5 also features an enhanced Contacts table to monitor favorable ligand-protein interactions:
- Interactive: clicking on a ligand/protein atom label in the Contacts table highlights the atom in the 3D window, double-clicking focuses the 3D view on that atom
- Table creation is significantly faster
- The table is now saved within the Flare project and can be exported
Other enhancements include improved support to peptide ligands and support for post-translationally modified amino acids.
Generating sensible binding poses for your ligands
We have significantly expanded Flare’s capabilities to generate a sensible binding hypothesis for your ligand series with the inclusion of FieldTemplater™ accurate field pharmacophores (Figure 7).
FieldTemplater enables you to work in 3D, modeling ligand properties such as electrostatic and shape, even when protein structure information is not available for your target. In cases such as GPCRs, ion channels and novel targets, FieldTemplater is the best way to reconstruct the bioactive conformation of the ligands, giving simple and easy to interpret results.
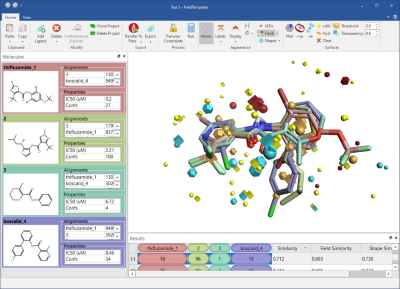
Figure 7: FieldTemplater accurate field pharmacophores enable you to work in 3D even when protein structure information is not available for your target.
Docking and scoring in Flare V5 using the LeadFinder™ algorithm features an expanded choice of docking constraints. You can apply these constraints to protein atoms/residues to ensure that ligand poses match relevant protein-ligand interactions. The full list now includes:
- H-bond forming protein atoms
- Metals
- Cation-pi interactions
- Aromatic-aromatic interactions
- Salt-bridge interactions
Finally, all advanced options for ligand-based alignment from Forge™ are now available in Flare together with the Conformation Explorer to inspect and analyze conformation populations (Figure 8).
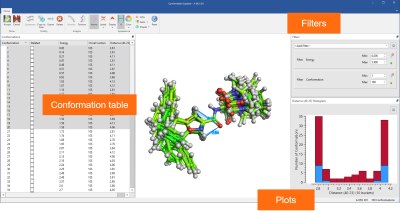
Figure 8: Use the Conformation Explorer to inspect and analyze conformation populations generated in Flare.
Powered by the Flare Python API
A Python API has been available for Flare from the earliest releases, and has constantly improved across the years to give you full access to all Cresset methods. As part of the integration of Forge into Flare, all ligand-based methods are accessible from the command-line using a single pyflare binary and Cresset-developed python scripts which are directly installed with the software.
To facilitate the transition from Forge to Flare, the python scripts use command-line options which are identical to those of the original Forge binaries.
This significantly simplifies the creation of integrated workflows. In Figure 9 - left, we show a Forge workflow to generate QSAR models to predict the activity of new compounds and understand their SAR. In Figure 8 – right, the same workflow can be simplifying by writing a single python script which performs all necessary steps.
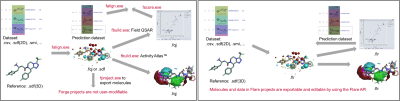
Figure 9: Left: A Forge workflow to generate QSAR models to predict the activity of new compounds and understand their SAR. Right: The same workflow can be simplified in Flare writing a single Python script which performs all the necessary steps.
Flexible licensing and free evaluation
Whether computational chemist, medicinal chemist, or academic, there is a licensing option to suit you. Try Flare V5 on your projects – request an evaluation.



















