Flare™ V4 release: Smarter and faster FEP calculations, new and improved force fields, improved Dynamics analysis tools

FEP calculations: Smarter and faster
Everybody loves FEP, a method for predicting relative binding affinity changes within a congeneric ligand series, because it can give very accurate predictions. At the same time, everybody hates FEP because of the long calculations, which require dedicated GPU resources.
After successfully demonstrating (Assessment of Binding Affinity via Alchemical Free Energy Calculations J. Chem. Inf. Model. 2020) that the implementation of FEP in Flare has a very good predictive ability on standard datasets, this release focuses on improving its calculation performance. At the same time, different types of perturbation networks and calculations modes are available to help you find your own optimal balance between speed and precision.
Let’s start with the high-precision solution. Fully connected, two-ways networks (figure 1) are the most precise type of FEP perturbation networks. Automatically built by Flare, they include additional transformations to form cycles between three or more ligands. This introduces some redundancy to the network, enabling more accurate error estimate for each link and thorough analysis of cycle errors.
Furthermore, the new ‘Production’ mode uses available experimental affinity values to optimize the network by ensuring that each new ligand to predict is never too far away (in terms of number of FEP transformations) from a ligand with known activity. This feature, together with an optimized algorithm for calculating predicted affinity values and associated errors, leads to considerably more precise calculations, which are also slightly faster, as links connecting compounds with known affinity will not be calculated.
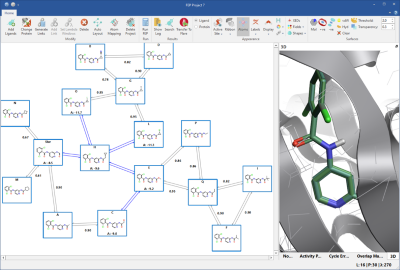
Figure 1: Fully connected networks introduce some redundancy to the network, enabling more precise calculations. In production mode, the blue links connect ligands with known experimental affinity, and will not be calculated.
If you are interested in exploring how changes to a single compound affect activity, you will like the new ‘star graph’ networks (figure 2) which are ideal for lead optimization projects. In this type of network, the selected, parent ligand molecule sits at the center of the star, with the molecules you want to predict shown at the edges. These networks involve a smaller number of links/transformations, so are significantly faster than more connected networks.
For both types of network, a new ‘quick mode’ is available, which will calculate each link in the perturbation network in one direction only, leading to 50% faster calculations.
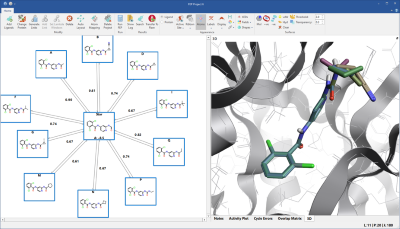
Figure 2. ‘Star graph’ networks are ideal for the optimization of a single lead compound, shown in the centre of the network.
In terms of speed, all FEP calculation in Flare V4 are significantly faster thanks to an improved algorithm with a 20% increase in performance, and they are fully parallelizable. This means that each bit of the transformation of one compound into another (the lambda windows) can be run on separate GPUs: the results will be merged at the end of the calculation. This can bring the calculation time for a single transformation on a medium-sized protein to less than 2 hours on a small cluster of 10 GPUs (for example AWS g4dn.xlarge, Tesla T4 spot instances). If you want to quickly make predictions for many compounds, you just have to ensure you have enough GPUs.
If your concern is the cost of GPU resources, then we have good news for you, as these don’t have to be too expensive. Our implementation of FEP can happily and robustly run on GPUs of gaming quality (such as GTX1080 or GTX2080), as well as on a variety of cost-effective Amazon spot instances. You can also decide for a mixed solution, for example maintaining a small cluster of GPUs in house, and renting out an AWS cluster only when needed, leaving it to the Cresset Engine Broker to guarantee a seamless connection to all GPU resources, both in-house and on the cloud.
New and improved force fields for FEP and Dynamics calculations
Quality of force field parameters for small molecule ligands is a key factor in the success of FEP and Molecular Dynamics calculation.
This release expands the choice of force fields available for this type of calculations with the addition of the Open force field. As the Open FF Consortium provides frequent updates and improvements to the Open FF, we opted for a flexible implementation within Flare, enabling you to easily upgrade to the latest available version simply by dropping the related files into the appropriate Flare installation folder.
If you choose to use AMBER/GAFF and AMBER/GAFF2 parameters for small molecules, the new Custom Force Field wizard provides a user-friendly interface to the calculation of improved custom parameters using the psi4 quantum mechanics engine. In the wizard, you choose the torsion for which you want to calculate new parameters, trim down your ligand to a representative fragment (figure 3 – left) to reduce the duration and noise in the calculation, choose the desired level of theory (figure 3 - right) and you are ready to start the calculation.
The results will be reported in the Custom Force Field Parameter explorer (figure 4) and saved locally to be used for FEP and Dynamics calculations.
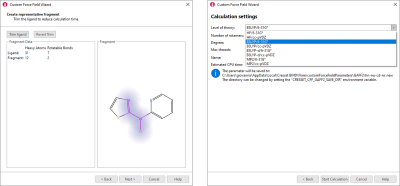
Figure 3. Development of custom AMBER/GAFF2 and AMBER/GAFF parameters is made simple by the Custom Force Field wizard.
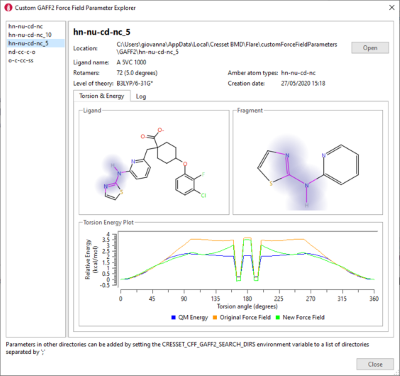
Figure 4. Custom Force Field Parameter explorer.
Dynamics analysis tools
Molecular Dynamics simulations can be run in Flare using OpenMM, to study the conformational changes of proteins and assess the stability of protein-ligand complexes. In Flare V4, these experiments benefit from the availability of new and improved force fields (see above) and are enriched by new visual tools for the analysis of the results. These include new interactive plots of chosen system properties (Potential Energy, Temperature, Box Volume and Density), the new table of Contacts (top-left corner in Figure 5), reporting contact statistics for favorable ligand-protein interactions observed during the simulation, and the creation of ad-hoc plots for any measurement (distance, angle or torsion) shown in the 3D window (figure 5 – right).
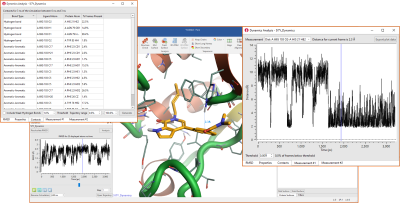
Figure 5. New visual tools for the analysis of Dynamics simulations in Flare V4.
Additional new features and improvements for medicinal and computational chemists
Many of the following new features and improvements stem from customer requests – so if there is something else you would like to see, let me know.
- Structure Check function, to check protein structures for potential problems which could not be resolved during protein preparation
- New advanced options for protein preparation, including the option to keep the ligand unchanged and the option to fix 1- and 2- residues gaps
- Improved ligand minimization, to minimize multiple ligands at the same time
- Improved handling of docking constraints
- New and improved GUI features:
- Draw protein surfaces as wireframe
- Color ligand and protein atoms by XED partial charge
- Export of ligands together with the associated protein
- Color Sequences function to change the background color of residues in the Proteins and Alignment tables to match the color shown in the 3D window
Flexible licensing and free evaluation
Whether computational chemist, medicinal chemist, or academic, there is a licensing option to suit you. Try Flare V4 on your projects – request a free evaluation.



















