The business case for outsourcing FEP calculations
Everyone seems to be talking about FEP: What is it and what do I need to know?
Flare™ V4 includes the ability to perform FEP calculations as a new addition to Cresset’s protein modeling suite (Assessment of Binding Affinity via Alchemical Free Energy Calculations J. Chem. Inf. Model. 2020). You may have heard several presentations from Cresset scientists on this topic at the recent Cresset User Group Meeting - don’t worry if you missed them as you can find presentations from the UGM here. If you are new to this area let me introduce you to FEP in Flare™ V4 , answer questions you may have regarding the technology, and illustrate how the Cresset Discovery Services team can assist with the application of FEP to your drug discovery projects.
Free Energy Perturbation methods represent the next step towards the goal of being able to predict the strength of the binding interaction between a small molecule ligand and a protein. These approaches are not new, however, they are now applicable to practical problems in drug discovery due advances in computer technology, mainly GPU array processors. The aim is to better define which of the many molecules that could be made are going to propel your project forward fastest.
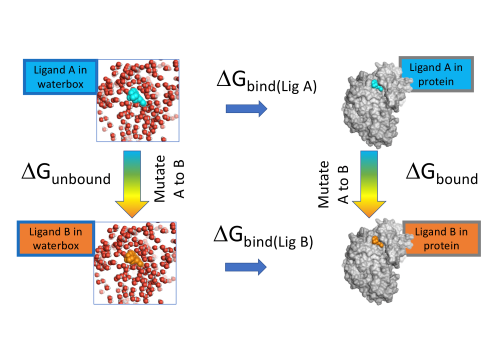
Figure 1: FEP in Flare calculates the difference in ΔG binding between Ligand A and Ligand B to a protein using alchemical changes to mutate A to B.
What are the challenges in the application of FEP to my drug discovery projects?
Many pharmaceutical companies have invested in in-house expertise and computational resources needed to apply FEP methods. Sosei Heptares, Janssen Pharmaceutica and Leiden University (working collaboratively) recently published a study which demonstrates the power of the application of FEP to drug discovery, with the caveat of the expertise required in application of the method. The Sosei Heptares collaborative study showed that using ‘out of the box’ parameters to set up calculations for GPCR targets did not deliver correlation with experimental affinity values. However, a series of logical steps, such as careful attention to positioning key water molecules, amino acid ionization states and in some cases additional equilibration, produced good results for sets of Adenosine A2 and Orexin OX2 antagonists. In the best cases the calculated values for ligand binding were within 0.6-0.7 kcal/mol of experiment for these important drug targets. These membrane-bound receptors are traditionally viewed as difficult targets to model, requiring bespoke treatment, as binding sites are often located deep within the receptor in comparison to soluble globular proteins.
This published example demonstrates the power of the application of the FEP method in the hands of experienced computational chemists. However, what if your company does not have an in-house computational chemistry team or a small computational chemistry team whose resources are already exhausted?
Why outsource FEP?
FEP methods involve a learning curve to achieve optimum results for drug discovery projects. Cresset Discovery Services offers the advantage of bringing to your projects a full range of scientific expertise in the application of the Flare™ V4 FEP method. From scientists who developed the method and software, to those who have tested and applied the method to practical drug discovery problems. Offering a full range of expertise from computational and physical chemists to medicinal chemists to software developers, our scientists work with you as part of your team, ensuring that the FEP method is applied efficiently and properly. This saves your team time, effort and energy and they can focus efforts on designing compounds rather than learning applications of computational methods and software.
Additionally, because we have our own (secure) in-house GPU cluster, there is no need for your company to invest in the purchase of additional specialized computational hardware resources. Current estimates are that one simulation takes roughly one day to run on a single GPU, the graphics chip that you have in your laptop. We use our in-house cluster of GPUs to reduce the time required for one simulation to a couple of hours per ligand/protein complex. Bear in mind that these are complex calculations and each target needs to be validated using data from a set of existing compounds (10-20) before predictions can be made. Ideally you will get the most accurate results if you calculate DG for both the change from compound A to new compound B and in reverse i.e., B to A, so a number of simulations are required before you can evaluate your own proposals. Not having to invest in your own GPU cluster available makes these types of calculations feasible.
Reduce bottlenecks
Bringing in external experts to assist your team with FEP can have a huge impact on hit-to-lead or fragment optimization. The actual bottleneck at this stage of a project is the limited chemistry resource, so that an ability to accurately rank custom-built libraries and identify the best candidates for synthesis is very valuable.
Expert CADD scientists for excellent results
Information required to begin the process
Setting up FEP calculations requires access to a structure of a representative ligand bound to your target protein –an ideal starting point would be an experimental X-ray structure. If this is not available, we can use their protein modeling expertise to generate carefully curated homology models, based on structures of closely related proteins, and calculate ligand binding poses using docking algorithms. The second requirement is a set of measured pKi values for 10-20 examples of your chemical series that span ~3 log units of activity.
Next steps
With this information in hand, FEP for your system is validated, configuring the calculation parameters and running a series of FEP calculations to check that the method reproduces experimental DG values for each compound, within a margin of 1.3 kcal/mol. Careful checks on both ligand and protein stability during the simulations are carried out and parameters optimized if appropriate. This stage requires significant computer resource, so sensible and efficient FEP experiments are performed and run on an in-house GPU cluster.
At this point your project will confidently move into production mode, using FEP to rank predicted biological activities of a bespoke library of new ideas, or the triaged results of a targeted virtual screen.
.png)
Figure 2: The individual steps we take to deploying free energy calculations in drug discovery projects.
Cresset Discovery Services offers expertise at each of these computational steps, from the complex initial setup, to homology modeling when necessary, to intelligence gathering for ligand structural and pki values. We can assist with the full calculation: with system preparation, generation and viewing of the perturbation map, viewing 3D structures and examination of the results and cycle errors, links where lamda windows don’t overlap can be easily identified, atom maps edited and new ligands added to an existing FEP study easily.
Conclusion
Adding Cresset Discovery Services computational expertise to your team will enable you to apply FEP for productive ligand design and advance your drug discovery projects efficiently and cost effectively.
Free confidential discussion
Reach out for a free confidential discussion to find out about the flexible service options for adding CADD expertise to your team.



















