Behind the scenes of Flare’s alchemical Free Energy Calculations
Last month, Giovanna Tedesco gave a sneak peek at some of the new features in the upcoming release of Flare™ V3, our structure-based design platform. Here I take a closer look at the main concepts and the underlying software of Flare’s FEP (Free Energy Perturbation) implementation.
FEP in a nutshell
FEP calculations are performed to predict the relative binding affinity changes (ΔΔG) within a congeneric ligand series. This is achieved by non-physical (‘alchemical’) transformations, in which a molecule (A) is gradually converted into a structurally related molecule (B) through a number of discrete steps, the so-called λ windows. The ligand simulated in each window can be thought of as an alchemical (i.e., hybrid) molecule consisting of a 1-λ fraction of A and a λ fraction of B (Figure 1).
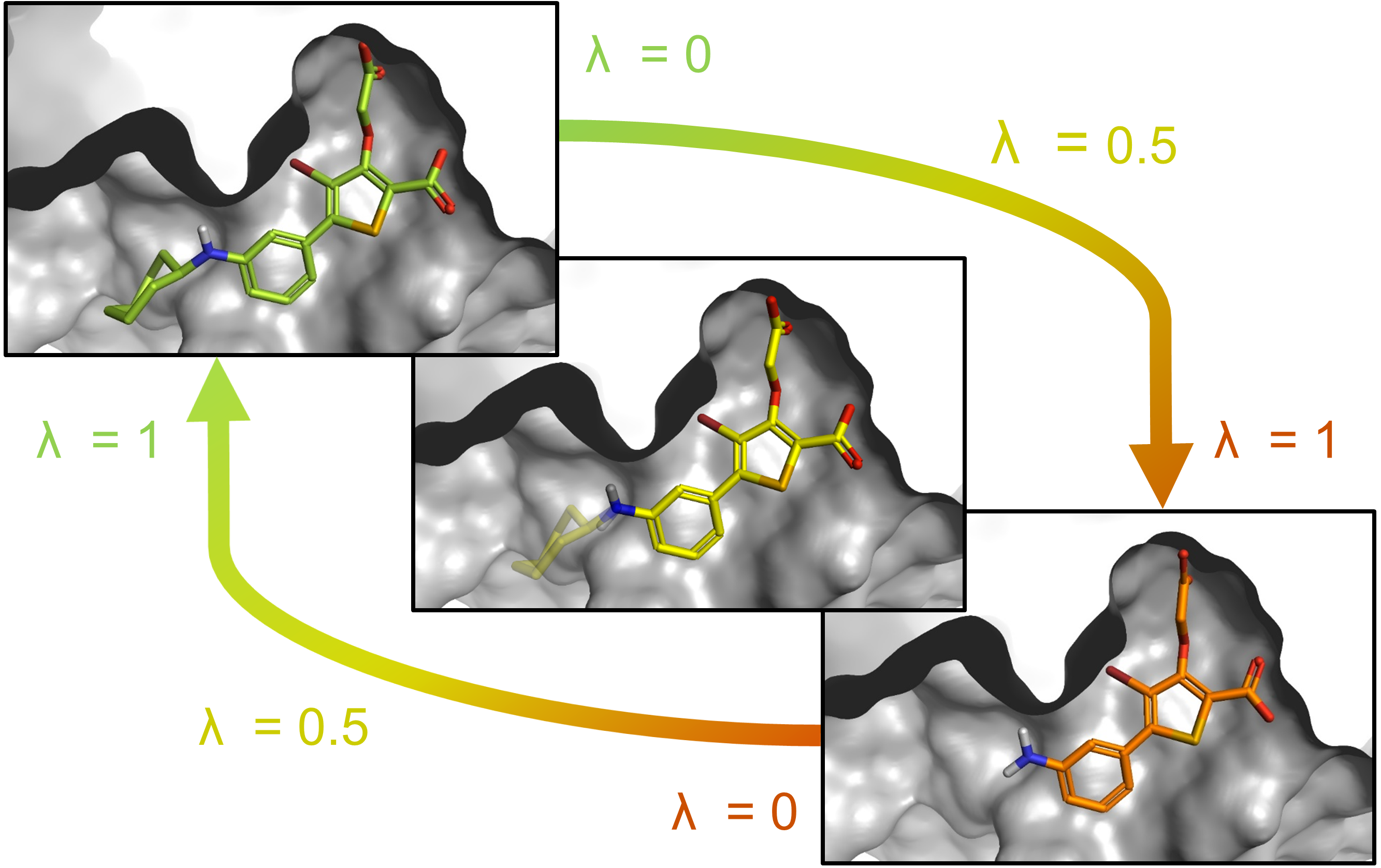
Figure 1: Ligands 20 (green) and 19 (orange) bound to PTP1B. An artificial hybrid molecule is shown in yellow. 20 has an additional cycloheptyl moiety.
The simulations are performed for each ligand pair of interest in the bound state (i.e., the protein environment) as well as in the solvated state (i.e., in a water box). The free energy difference between the end states of these transformations corresponds to the binding affinity difference between the two molecules: ΔΔG = ΔGbound - ΔGsolvated
How to set up an FEP calculation in Flare
Prerequisites to perform FEP calculations are a well-prepared protein structure and the binding pose of at least one molecule. Ideally, the binding poses and experimentally measured affinities of several molecules should be known, in order to compare calculated and experimental affinities. As accuracies of about 1 kcal/mol can be achieved via FEP, the experimental values should span at least two orders of magnitude. To generate the starting poses for the FEP calculations, congeneric ligands should be placed in the binding pocket using the Lead Finder™ template docking algorithm within Flare. Once the described protein and ligand structures are obtained, the actual work on FEP can commence.
The perturbation network
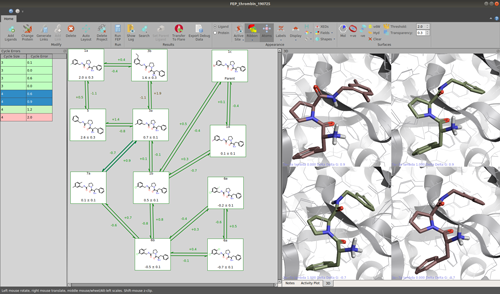
The first step when starting a new FEP project in Flare is the generation of a perturbation network. This is a crucial task, as performing calculation on pairs of structurally similar molecules is computationally less demanding and yields more reliable results. However, rather than just performing pairwise perturbations, it is recommended to include additional perturbations in order to form cycles between three (or more) ligands. This introduces some redundancy to the network, and thus enables more reliable error assessment.
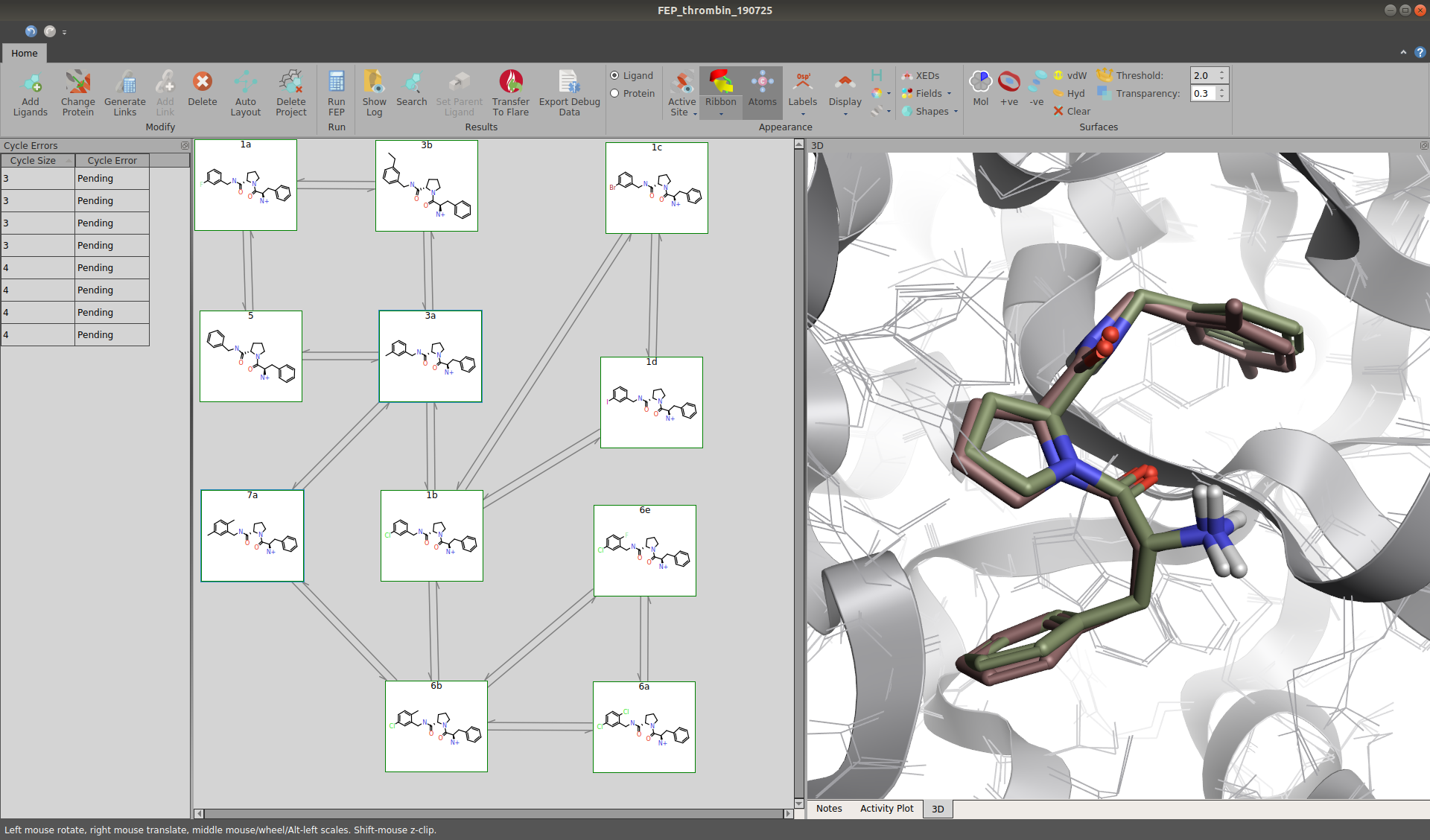
Figure 2: Network of a small data set of 11 Thrombin inhibitors.
As drafting a network manually is a tedious task, Flare handles this in a fully automated fashion by using an extensively revised version of the open source tool LOMAP[1]. Roughly speaking, every possible pair of molecules is scored based on their structural similarity, taking into account their orientation in the binding pocket and possible inversions of chiral centers. Afterwards, an interactive map of the network is generated, in which similar ligands are grouped together. There is also direct feedback to the user when connected ligand pairs are likely to give poor results. This enables the user to implement additional intermediate molecules to ensure reasonable perturbations before actually starting the computationally demanding molecular dynamics (MD) simulations. Furthermore, the network may be freely customized to allow for different degrees of redundancy. It is worth noting that additional molecules may always be added to an existing network while keeping the results of already performed calculations.
Single topology approach
This initial release of Flare V3 performs perturbations using the so-called single topology approach. This means that the molecules in the end states (λ = 0 or 1) share as many atoms as possible, and dummy atoms are introduced to account for any additional atoms. Direct transformations of two rings are only possible when they have the same number of atoms, to avoid inconsistencies in the parameters of the hybrid molecules. Hence, networks containing molecules with differently sized rings must include a common intermediate structure (e.g., three ligands such as: R–cyclopentyl ⇐⇒ R–H ⇐⇒ R–cyclohexyl, where R is the maximum common substructure). While this hinders experiments like the macrocyclization of ligands, scaffold/core hopping case studies are within reach of the method.
FEP calculations
Before starting the FEP calculations, several parameters like the simulation time and the number of λ windows can be freely adjusted. There must be an adequately high number of windows for each perturbation, as the potential energy difference between each pair of windows must be sufficiently small in order to obtain converged free energy differences (this ‘phase space overlap’ can be checked from overlap matrices obtained after the simulations have finished.) Hence, the number of windows may be set to different values, depending on the difficulty of each perturbation.
The remaining steps are carried out without any intervention by the user: input files for the MD simulations are generated using AmberTools[2] and a multi-step equilibration protocol is carried out using OpenMM[3] and the Amber ff14SB[4] and GAFF/GAFF2[5] force fields. Several software packages implemented or built on top of the Sire[6] framework are used to carry out and analyze the actual FEP calculations: input files are generated using BioSimSpace[7] and the simulations are performed by SOMD. Once these have finished, the potentials of mean force for each perturbation are calculated using the MBAR algorithm[8] and, finally, the ΔΔG values are calculated.
Results and reliability
Before comparing the computed free energies to experimental values, the reliability of the calculations should be assessed. Several easily accessible ways are available in Flare to do so: For instance, the structures at the beginning and the end of the simulations can be easily checked to ensure that the final binding poses are reasonable.
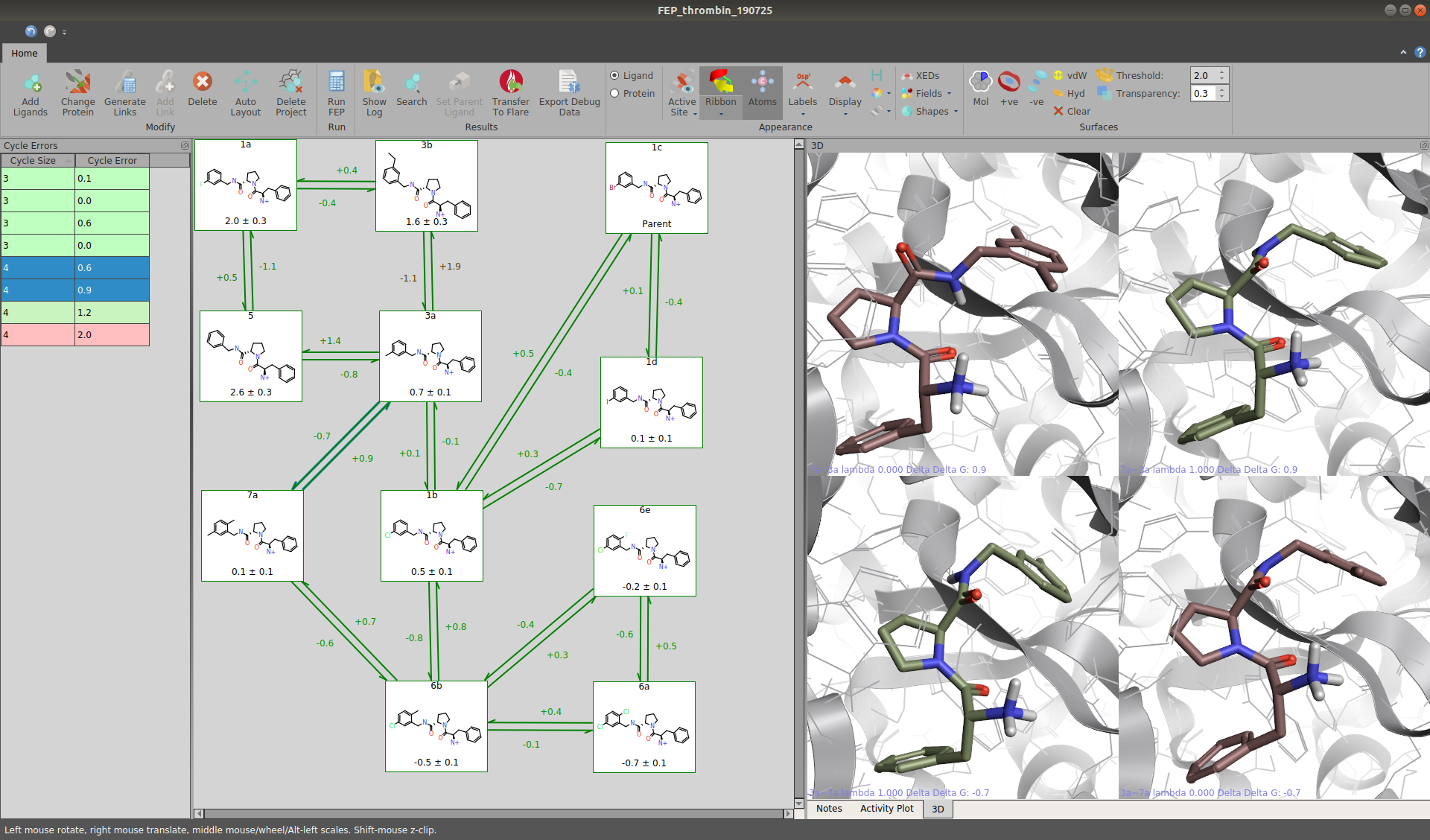
Figure 3. The same data set as in figure 2, but after the calculations have finished. Structures at the beginning and the end of each perturbation can be viewed on the right. Hysteresis can be assessed directly from the network and the cycle errors. These are shown in the table on the left-hand side. Questionable results are highlighted in red.
Furthermore, it should be checked that the hysteresis (i.e., the difference between the energy calculated when transforming molecule A to B and the calculated energy of transforming B to A) for each ligand pair is fairly low (ideally, it should be zero). Inaccuracies of the ΔΔG values in cycles of the perturbation network may also be assessed. Both checks can be performed quickly in the interface, as issues are highlighted according to an intuitive color scheme. Questionable perturbations may be rerun or simply excluded from the final analysis. To ensure that there is sufficient phase space overlap between the λ windows, users may also refer to the reported overlap matrices. A warning is provided if the overlap is not at least at least 0.03, which signals that the simulation may not have sufficient λ windows to provide accurate results[9].
Computational resources
As GPUs are needed to perform the FEP calculations, the Cresset Engine Broker enables Flare to connect to a local cluster as well as to cloud computing facilities with minimal setup time for the user.
Internal benchmarks suggest that a single uncomplicated perturbation with only 9 λ windows and a medium sized protein (JNK1, 358 residues) can be carried out in about 30 hours on a single modern GPU. Accordingly, a decently sized dataset consisting of 30 ligands and 42 perturbations can be calculated on a small cluster with 16 GPUs in under 7 days. More GPU resources enables users to assess more ligands or obtain shorter calculation times are easily.
Conclusion
The implementation of FEP in Flare builds on top of the best open-source tools and combines them with Cresset’s expertise in delivering easy-to-use software. The intention is to provide our users with an accessible and user-friendly interface to FEP, using a fully automated workflow internally validated using different datasets. At the same time, full control over the simulation parameters both within Flare and through the Flare Python API facilitates the exploration and identification of the ideal conditions for a given set of ligands and their target protein.
Book your free evaluation
FEP will be available in the upcoming release of Flare V3; book your free evaluation now.
References
- Liu, S., Wu, Y., Lin, T. et al. J. Comput. Aided Mol. Des., 2013, 27, 9 755-770
- AmberTools
- OpenMM
- Maier, J. A., Martinzes, C., Kasavajhala, K. Et al. J. Chem. Theory Comput., 2015, 11, 8, 3696-3713
- Wang, J., Wolf, R. M., Caldwell, J. W. et al., J. Comput. Chem., 2004, 25, 9, 1157-1174
- SireMol
- BioSimSpace
- Shirts, M. R., Chodera, J. D., J. Chem. Phys., 2008, 129, 12, 124105
- Klimovich, P. V., Shirts, M. R., Mobley, D. L., J. Comput. Aided Mol. Des., 2015, 29, 5, 397-411



















