Do Structures of the Coronavirus Spike Protein Hold the Key to Winning?
In this blog Peak Proteins spotlight the homotrimeric spike (S) glycoprotein of the novel coronavirus, 2019-nCoV, and what is currently known at the molecular level about how the virus operates.
The global covid-19 pandemic is at the forefront of most people’s minds at the moment, and there has been a monumental effort in the scientific community to try and understand every small detail of the enemy down to the last detail. In this blog we spotlight the homotrimeric spike (S) glycoprotein of the novel coronavirus, 2019-nCoV, and what is currently known at the molecular level about how the virus operates.
A sugary molecular crown
The S protein protrudes from the viral membrane, and its characteristic ‘crown’ shape gives this class of viruses their name. In the months since the start of the year, a number of different research groups have managed to solve the three-dimensional structures of parts of the S protein both by X-ray crystallography and cryoEM. Normally, such technically challenging research doesn’t provide answers so quickly – the time between discovering the protein and solving the structure in not just one, but multiple different states is record breaking. Aside from the global need to combat the virus, the speed at which the results were obtained can be attributed to a combination of collaborative community-based research and borrowing prior tricks from working on SARS-CoV, a highly similar virus that caused the SARS outbreak in 2002.
CryoEM structures from Wrapp et al. 2020 and Walls et al. 2020 have afforded us our first glimpse of the 2019-nCoV S protein, and largely, it is the same as its counterpart in SARS-CoV, unsurprising as it shares 98% sequence identity. Three copies of the S protein form an intertwined complex, each copy having an S1 domain forming the outer part of the structure, and the S2 domain that forms the inner core and terminates in a membrane spanning region to anchor the complex on the viral membrane (Figure 1A and B). The S1 domain contains the receptor binding domain (RBD), which is the part of the S protein that recognises and binds the target on the host cell membrane. Furthermore, the outside of the S protein complex is decorated with a complicated pattern of glycosylation, which forms a carbohydrate shield that disguises the virus from the host immune system – the flexible sugary chains are difficult for antibodies to recognise and bind to (Figure 1C, Grant et al. 2020).
Figure 1 – Structure of the trimeric 2019-nCoV spike glycoprotein (click here to view).
- A) top down view at the homotrimeric 2019-nCoV spike glycoprotein with separate chains highlighted in red, green and blue. B) Side view of the same representation as in panel A. The membrane spanning region was absent from constructs used for cryoEM studies, and therefore could not be visualised. However, the relative location of the membrane spanning region has been indicated. C) Positions of N-glycosylation attachment sites on the trimeric S protein as indicated in the cryoEM structure. Full glycosylation chains could not be visualised as they are too flexible and thus were averaged (‘blurred’) out of the structures. The cryoEM model with the PDB code 6VXX (Walls et al. 2020) was used for this visualisation.
The prefusion to postfusion transition
The cryoEM structure of the S protein in an open conformation (Walls et al. 2020), a crystal structure of the RBD domain bound to the peptidase domain (PD) of the second isoform of human angiotensin-converting enzyme 2 (ACE2) (Wang et al. 2020), and a cryoEM structure of an ACE2/sodium-dependent neutral amino acid transporter (B0AT1) bound by an RBD domain (Yan et al. 2020) provide a snapshot of the first step in viral recognition at the membrane. The S protein is usually in a compact closed form, but in the presence of the ACE2 target on the host membrane, one copy of the S1 domain undergoes a large conformational change to an open conformation in order for the RBD to engage with the target (Figure 2). Following recognition at the membrane, this open prefusion complex is then cleaved into the separate S1 and S2 subunits. The S1 subunits are released and the complex transitions to the postfusion conformation where the S2 domain undergoes its own shape change (PDB 6LXT, Xia et al. 2020), allowing it to orchestrate fusion with the host membrane.
Figure 2 – A model of the prefusion S protein complex bound to ACE2 at the cell membrane (click here to view)
This visualisation was made by overlaying structures of the open trimeric S protein with PDB codes 6VYB (Walls et al. 2020) and 6VSB (Wrapp et al. 2020), the crystal structure of the ACE2 PD domain bound by the RBD domain of the S protein with the PDB code 6M0J (Wang et al. 2020), and the structure of the B0AT1/ACE2 complex bound to the RBD domain of the S protein with the PDB code 6M18 (Yan et al. 2020).
Why is 2019-nCoV more virulent than SARS-CoV, despite their similarity?
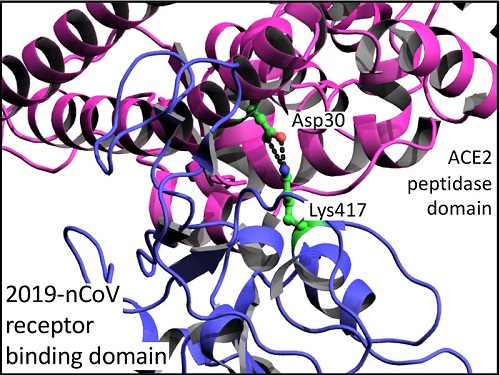
There are a number of subtle differences in amino acid sequence of S proteins that may help explain why the 2019-nCoV RBD has a 10 to 20-fold higher affinity for ACE2 than SARS-CoV RBD. Amongst a number of smaller differences, there is a marked difference of an additional salt-bridge interaction between Lys417 of the 2019-nCoV RBD and Asp30 of ACE2 that is absent in SARS-CoV RBD (Figure 2; Wang et al. 2020). However, Wang et al. cast doubt that these differences are the sole reason for 2019-nCoV increased virulence as, additionally, the 2019-nCoV S protein has a furin cleavage site (RRAR) at the S1/S2 boundary that is a single arginine residue in the SARS-CoV S protein. Interestingly, furin sites are also found at a related position in hemagglutinin proteins of highly virulent avian and human influenza viruses (Chen et al. 1998).
Can the 2019-nCoV S protein/ACE2 interaction help explain susceptibility?
The interaction between the S protein and ACE2 is clearly important in the process of viral infection, and it is possible that natural variation in the amino acid sequence of ACE2 amongst the human population has baring on susceptibility of individuals to covid-19. A number of computational studies have tried to address this point (Li et al. 2020, Renieri et al. 2020), and suggest that indeed natural variation of the ACE2 sequence may play a role in susceptibility and severity of individuals to 2019-nCoV infection.
Therefore, further study of these structures will be important as, taken together they provide a detailed narrative of the steps that lead to viral invasion of a host cell, and provide molecular level information about the characteristics of infection and susceptibility. Importantly, the information may hold clues as to how we might stop the process of viral invasion, either with small molecules or antibodies, a step towards halting the virus from spreading.



















