GPCRs: ‘you’ve gotta know how to hold them, you’ve gotta know how to fold them!’
There are challenges of working with a specific sub-type of membrane proteins called G-protein coupled receptors (GPCRs) however, it doesn’t have to be a labour-intensive process at all.
Walk the line
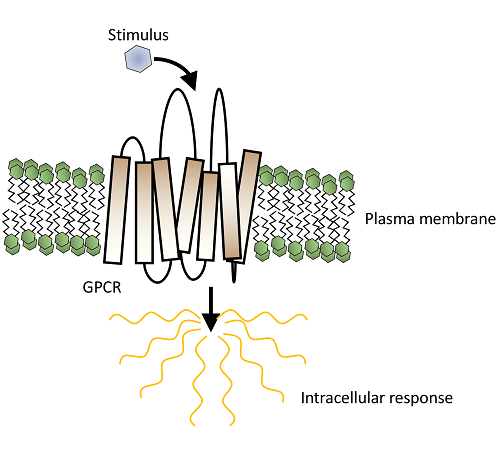
In one of our previous website blog posts we discussed the challenges of working with a specific sub-type of membrane proteins called G-protein coupled receptors (GPCRs). All GPCRs have seven transmembrane alpha-helices that span the plasma membrane, and flexible extra- and intracellular loops. The extra- and intracellular sides of a GPCR are responsible for signal detection on the outside of a cell and signal transduction inside the cell, respectively (Figure 1A). These helices are very hydrophobic, allowing them to traverse the plasma membrane, but means they are water insoluble. Therefore, there is a thin line to walk in GPCR purification where one has to balance the need to shield the hydrophobic environment of those helices and, at the same time, expose the protein to the aqueous environment of purification buffers. The process that aims to satisfy these two requirements is solubilisation, usually through the use of a mild detergent, such as n-Dodecyl-β-D-Maltoside (DDM) which replaces native lipid bilayer with a detergent micelle (Figure 1B).
Figure 1 (Click here to view). A) GPCR signaling. GPCR receives a signal from extracellular environment in form of a ligand, a change in pH or in a form of a physical force applied to the receptor. This causes conformational changes with GPCR itself and leads to the dissociation of a G-protein from the intracellular face of the receptor, and subsequent intracellular signaling. B) Solubilisation. Native plasma membrane phospholipids become replaces with a mild detergent (DDM in this case) and enclosed into micelles.
Major issues encountered when working with membrane proteins include low expression levels and poor stability. Over the years, different methods have been developed to overcome intrinsic instability of GPCRs. In our previous blog post we covered the use of fusion partners and introduction of stabilising amino acid point mutations. However, approaches that don’t require protein engineering are also available, including the optimisation of solubilisation and buffer components, such as; the addition of the cholesterol analogue, cholesteryl hemisuccinate (CHS) or a ligand that locks the GPCR in a stable conformation. Although these tricks are well known, in the effort to obtain GPCR crystal structures, the process can still be painful and needs to be carefully navigated, so gathering as much information regarding optimal conditions beforehand is crucial. At Peak Proteins we’ve looked at three well characterised GPCRs; A2A, S1P1R and 5HT2A and applied different techniques from low to medium throughput that allow systematic screening of optimal conditions for GPCR solubility, monodispersity and stability.
Don’t disperse and avoid aggregation
Purifying a badly behaving protein can often take a whole week, and there’s nothing more annoying than to find it’s crashed out of the solution by the end. However, a prerequisite for an informative screening assay doesn’t require pure protein at all; only a cleverly designed construct! All of our GPCRs were tagged with green fluorescent protein (GFP), allowing easy selective detection of very low concentrations of our protein with high sensitivity (Figure 2A). A technique that takes advantage of these properties is fluorescent size-exclusion chromatography (FSEC) (Kawate and Gouaux 2006), in which crude cell solubilates containing the GFP-tagged GPCR are passed down a size-exclusion chromatography (SEC) column, and fluorescence signal (rather than A280 absorbance) monitored to detect where the GPCR elutes (Figure 2B). We can then use handy metrics such as the monodispersity index (a ratio of the maximum absorption in the monodisperse peak vs. maximum absorption aggregation peak (Audet et al. 2019), or the relative magnitude of total signal to compare the effect of different solubilisation or buffer conditions. In its simplest form, FSEC doesn’t require expensive specialist equipment or reagents, and can be run using a basic ÄKTAprime, a 24 mL SEC column and a fluorescent plate reader (as we do). This provides a medium throughput workflow (you can do five screens a day if you’re organised!) and provides ample information regarding protein solubilisation and stabilisation. However, with a HPLC system equipped with an autosampler, an analytical SEC column and an online fluorescence detector, FSEC can be taken into high-throughput mode!
Figure 2 (Click here to view) A) GPCR constructs used in solubilisation screening conditions. B) Fluorescent size-exclusion chromatography.1. Solubilise cells with detergent; 2. Low-speed spin; 3. High-speed spin; 4. Supernatant loaded a SEC column; 5. Fractions collected; 6. GFP-fluorescence detected using plate reader. C) GFP-based thermostability assay.
Don’t melt down
The GFP-tag can also be used in GFP-based thermostability assay, where crude lysate containing the GFP-tagged GPCR is treated with a temperature gradient followed by centrifugation to remove heat-denatured protein. Fluorescent signal remaining in solution indicates the proportion of receptor that is heat-resistant relative to denatured, and allows recording of a melting-curve, the mid-point of which is taken as the apparent melting temperature (Tm; Figure 2C). We used our example GPCRs to observe shifts in Tm in response to different ligands (Nji et al. 2018). With this quick assay we were able to ascertain that some antagonists specific for a particular GPCR caused a reduction in Tm, perhaps locking them in a conformation that is destabilising, despite a wide consensus that these ligands are stabilising in nature. On the other hand, ligands that are supposedly non-specific for a receptor may still bind non-conventionally and exert allosteric effects that stabilise the receptor, as we found that surprisingly, risperidone (a 5HT2A antagonist) showed a stabilising effect on all three of the GPCRs, not just 5HT2A! This only served to confirm that finding optimal conditions for protein stabilisation is not straightforward or even intuitive.
Although FSEC and GFP thermostability are rapid and have low sample requirement, they neglect the fact that a protein could be interacting with a myriad of chaperone proteins in the lysate that may have stabilising effects, an occurrence almost never seen in biophysical or structural studies. Thus, an alternative thermofluor stability assay can be used on highly purified protein samples (see our blog entry here). In our case, we utilised thiol-binding dye 7-Diethylamino-3-(4′-Maleimidylphenyl)-4-Methylcoumarin (CPM) that emits fluorescence upon reacting with free thiols on cysteine residues as the protein unfolds due to temperatures (Alexandrov et al. 2008, Harborne et al. 2017). Although a bit more labour intensive this assay can provide precious information regarding the stability for a low amount of protein required.
Sometimes the task of optimising conditions for unstable proteins may seem daunting; however, it doesn’t have to be a labour-intensive process at all. With the right construct design, there is a plethora of assays that can inform on protein stability under different conditions.
To find out more about how we tackle difficult proteins have a look at our Case Studies!




















{kind=link}
{kind=link}