Sneak peek at Flare V3: New science and improved results
As we move closer to the release of Flare™ V3, let’s have a quick look at some interesting new functionality in this release.
Free Energy Perturbation
Free Energy Perturbation (FEP) calculations allow for prediction of relative binding affinity changes within a congeneric ligand series. In these calculations a molecule is gradually converted into a structurally closely related analogue via a non-physical (‘alchemical’) pathway. By assessing the free energy difference (ΔΔG) between the end states of such transformations, accuracies of about 1 kcal/mol compared to experimental values can be achieved for large datasets.
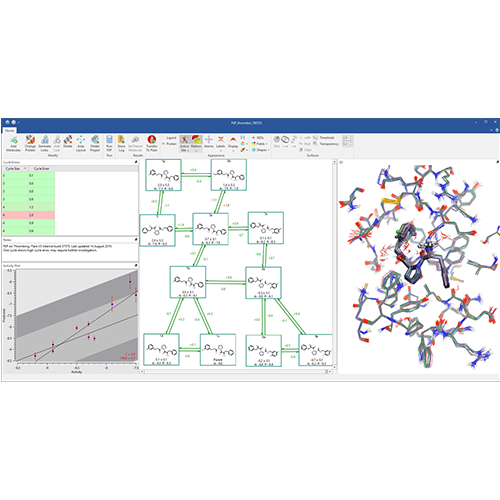
Implementation of FEP in Flare V3 combines open-source tools such as AMBER tools, OpenMM, LOMAP, Sire and BioSimSpace with Cresset’s expertise in delivering intuitive software to deliver an accessible and user friendly interface to a fully automated FEP workflow (Figure 1).
.png)
Figure 1: The fully automated FEP workflow in Flare V3.
The FEP workflow in Flare has been internally validated on various datasets, including the FEP+ dataset as a reference benchmark. Full control over simulation parameters, both within the Flare graphical user interface (GUI) and through the Flare Python API, enables you to explore and identify the ideal conditions for a given set of ligands and their target protein.
Template docking
As Lead Finder™ (the docking algorithm in Flare) only explores the torsional space of the ligands, the choice of an appropriate 3D starting conformation may have a strong influence on the results of a docking experiment. Manual curation of the 3D starting conformation is, however, a tedious exercise, especially when working with a large number of ligands to dock.
Template docking in Flare V3 offers you a practical solution to the problem for those docking experiments where you know the pose of a ‘template’ ligand, and wish to use this information to bias the docking results for congeneric compounds. When using template docking, the molecules to be docked are aligned by substructure to the template ligand, and the aligned conformation is used to seed the docking run, generally leading to improved results as shown in Figure 2.
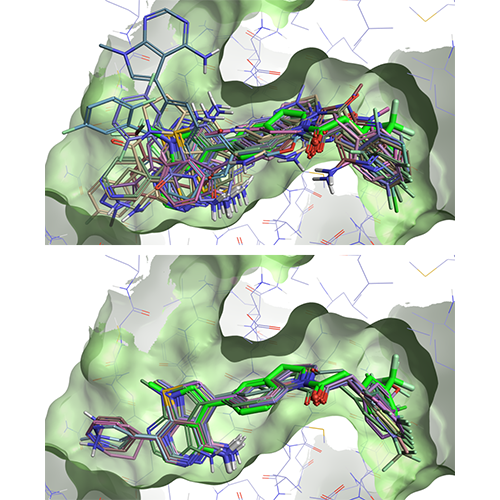
Figure 2: Left: docking of a set of 25 ligands in the PERK active site (PDB: 4G31). Right: template docking results for the same set of ligands, using the 4G31 ligand (bright green) as a template.
Additional features in Flare V3
When Flare V3 is released in the next couple of months you will also benefit from enhancements to molecular dynamics, covalent docking and ligand-based alignment, plus integration with the Blaze™ virtual screening platform.
Try Cresset solutions on your project
Try Cresset solutions on your project by requesting a free evaluation of any desktop application.



















