Driving Optimization of Antibiotic Drug Design

Since its heyday in the 1950s, the discovery of new classes of antibiotics has decreased, dropping to zero in the 1990s and not recovering since the turn of the century (Figure 1). Worryingly, this decline in novel antibiotic classes has coincided with the acceleration of antibiotic-resistant pathogens, meaning the current arsenal of treatments will be insufficient to deal with the demands that bacterial infections will pose in the future.1 Therefore, the requirement has never been greater to develop novel classes of antibiotics.
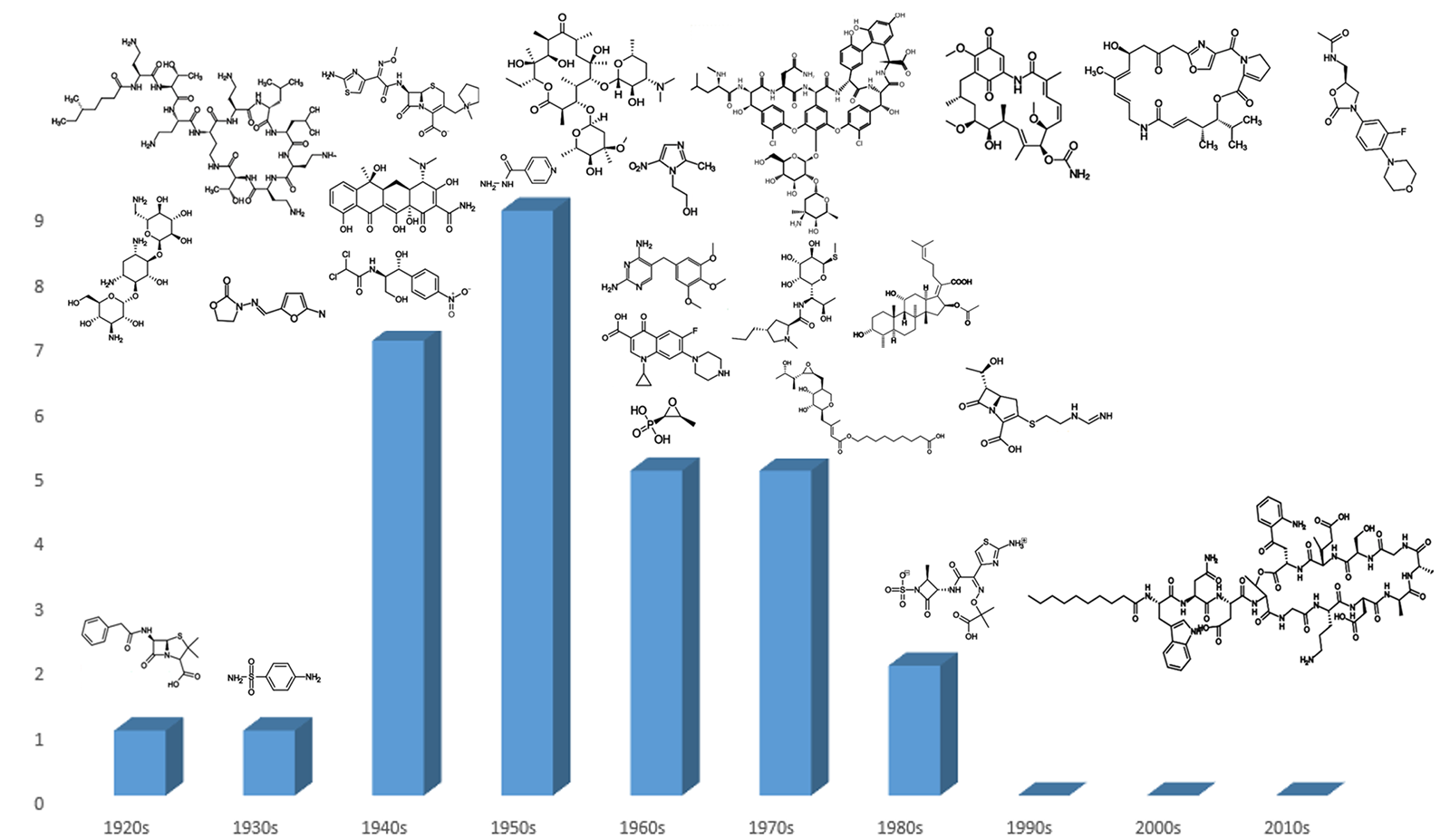
Figure 1. Classes of antibiotics arranged by decade of discovery.
Current Challenges: Potential Opportunities
As drug discovery processes have evolved, we have moved away from serendipitous discoveries, such as Fleming’s discovery of penicillin in 1928, being a common method of identifying new drugs. With the development of combinatorial and diversity-orientated methods of compound preparation and high throughput screening (HTS) platforms, much of the emphasis of hit identification has been on access to large numbers of compounds. A consequence of this juxtaposition is the chemotypes that chemists are working on has shifted from natural products and their analogues, which provided a rich source of novel antibacterial classes, to more synthetically tractable scaffolds with suitable functionality to rapidly explore several vectors.2
Pharmaceutical companies’ screening collections have been curated, at great expense, to maximize the probability of identifying compounds that can be developed into drugs to treat a variety of conditions. In an analysis by AstraZeneca, they showed that the average LogD of the compounds in their HTS collection (~2.5) was higher than that required to identify active antibiotics. Furthermore, it was demonstrated that this discrepancy was exacerbated when moving from gram positive to gram negative bacteria.3
Application of Novel Tools to this Challenge
While the development of novel antibiotics is undoubtably a challenge, novel computational technologies and approaches do offer complementary strategies that can be used to harvest valuable insights to progress drug discovery projects in this arena.
Due to the higher costs associated with both isolating and synthesizing natural products and their analogues, this area of chemical space has predominately been replaced by more cost-effective synthetic alternatives. The application of ligand-based virtual screening platforms allows screening of a higher number of compounds, thus greater coverage of chemical space, at a fraction of the cost of curating, analyzing, and maintaining a physical HTS collection. For example Cresset’s Blaze4 contains a database of 22 million commercially available compounds that can be accessed. Additionally, of course, the cost associated with running the HTS itself is also more expensive than conducting virtual screening. Cresset Discovery is applying this in silico methodology as part of a multi-year project in collaboration with the Summers group in the Department of Genetics at the University of Cambridge, on the development of a potential novel therapy for urinary tract infections.
Analyzing changing binding states
Many antibiotics work via a covalent mechanism of inhibition. This means that while most of the structural information that is available for this type of ligand that is published in the Protein Data Bank (PDB)5 will be in the covalent bound state, this is not the only molecule state that needs to be considered when modeling these ligand-protein complex systems. To illustrate this, Cresset’s FlareTM solution, for both ligand- and structure-based drug design6, was used to analyze and assess molecules in both their non-covalently bound ground state and their irreversible covalently bound state (Figure 2).
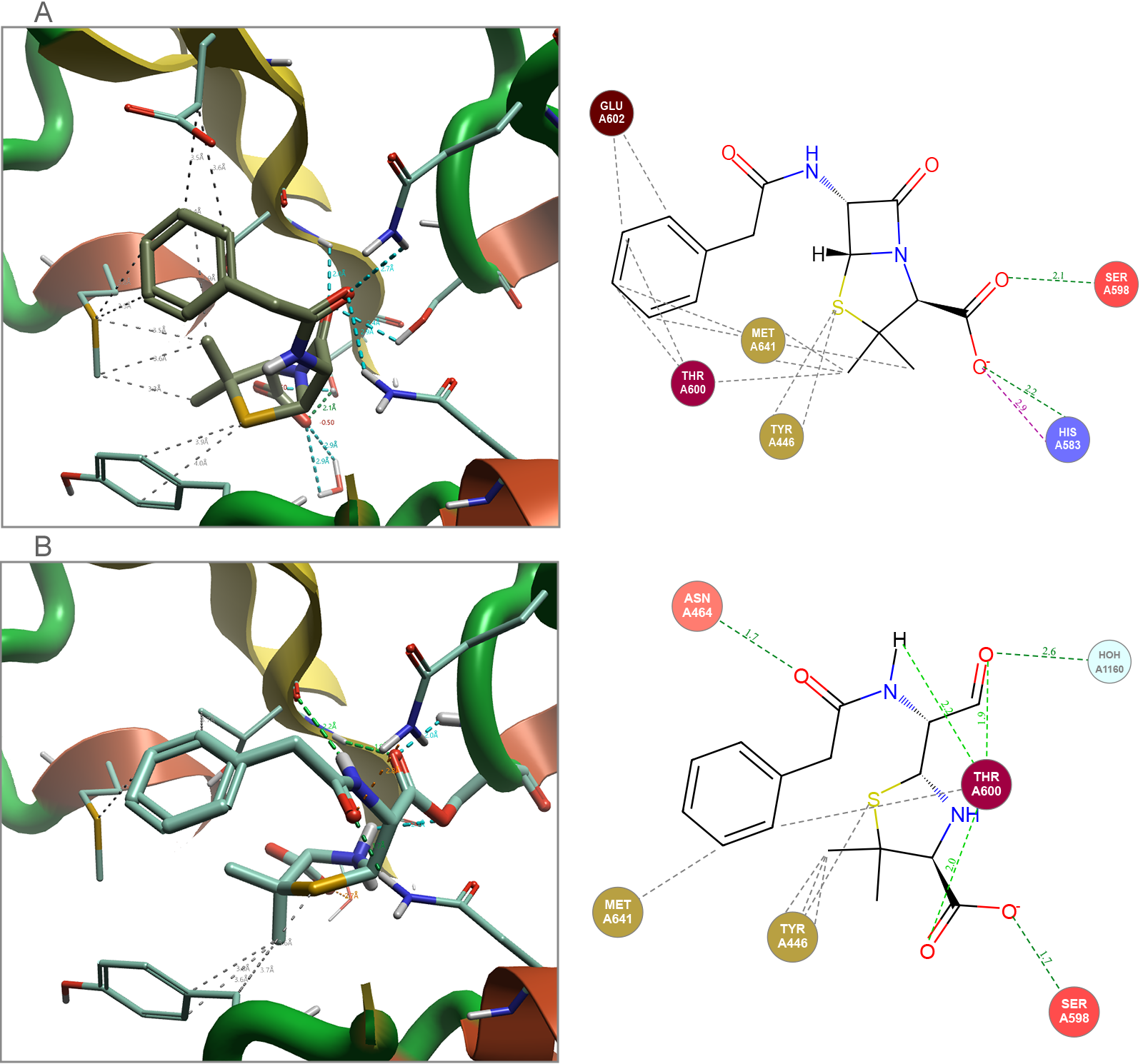
Figure 2. Penicillin G bound to Staphylococcus aureus (1MWT). (A) Docked non-covalent binding of the ground state. (B) Covalent binding of final complex. Images generated by Flare.
The non-covalent ground state ligand is the species that the protein encounters initially. Therefore, it is important that there is a strong binding event between this ligand and the protein, if this doesn’t occur any observed activity would result purely from covalent reactivity of the warhead, leading to lack of specificity and potential toxicity. The covalent bound ligand in the final complex allows further analysis of the binding process with respect to the orientation of the ligand in the protein, the covalent reaction of the β-lactam with the threonine OH group requires a release of ring strain within the ligand.
Navigating ligand-transporter interactions
The challenge of developing novel antibiotics is not limited to optimizing ligand binding to a particular target. In addition to these drugs requiring broad spectrum activity against a variety of bacterial species, a bastion of influx and efflux transporters must also be navigated to ensure that the drug has the required profile to allow binding to the required binding site. Due to the transient nature of ligand-transporter interactions, there are few examples of these complexes available in the PDB. Of the few that are available, there is little information that can be gleaned on the position of the ligand within the transporter, as these molecules are not binding to the same region of the protein (Figure 3).
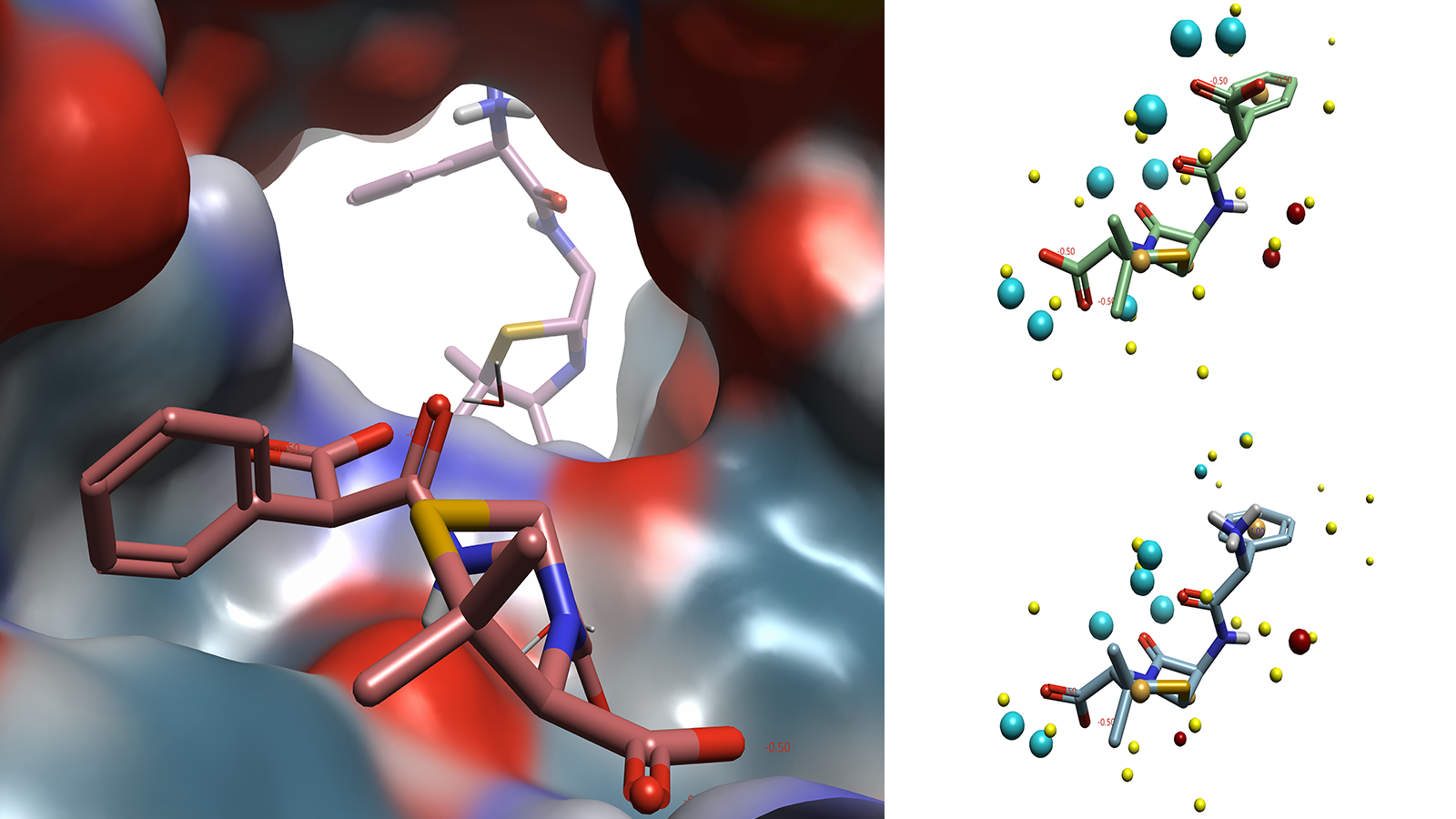
Figure 3. Carbenicillin (foreground) in E. Coli OmpF transporter(4GCQ) with ampicillin (background) from E. Coli OmpF transporter(4GCP) superimposed. Carbenicillin (top right) and ampicillin (bottom right) in conformation reported in crystal structure with field points added. Images generated by Flare.
Despite the lack of meaningful structural information on how these ligands interact with the transporter protein, the Cresset Discovery team can apply ligand-based techniques to gain valuable insights into a system. By utilizing specialist tools and applying their medicinal chemistry expertise, the team can map the ligands, which have no information on their biologically relevant conformation, against other ligands that display the same profile against a target. By applying Cresset technology used in SparkTM, Cresset Discovery's modelers apply their deep computational knowledge to carry out scaffold hopping and bioisosteric replacements7, or in Blaze, for virtual screening.
Generating new analogues through bioisosteric replacement
For example, the penicillin G analogues carbenicillin (with an additional carboxylic acid on side chain) and ampicillin (with an additional amine group on side chain) serves as the basis for illustrating an approach typically performed by Cresset Discovery's team, to model the conformation shown to pass through the OmpF transporter as a reference. A Spark search can be conducted to generate new analogues with alternative side chains that match the 3D field points generated for these molecules shown in Figure 3. The 500 ligands generated in Spark can then be independently docked into the staphylococcus aureus (1MWT) crystal structure described in Figure 2. In addition to the docking score generated, the Electrostatic Complementarity (EC)8 score can also be calculated as an additional metric to assess how well the molecule matches the protein of interest (Figure 4).
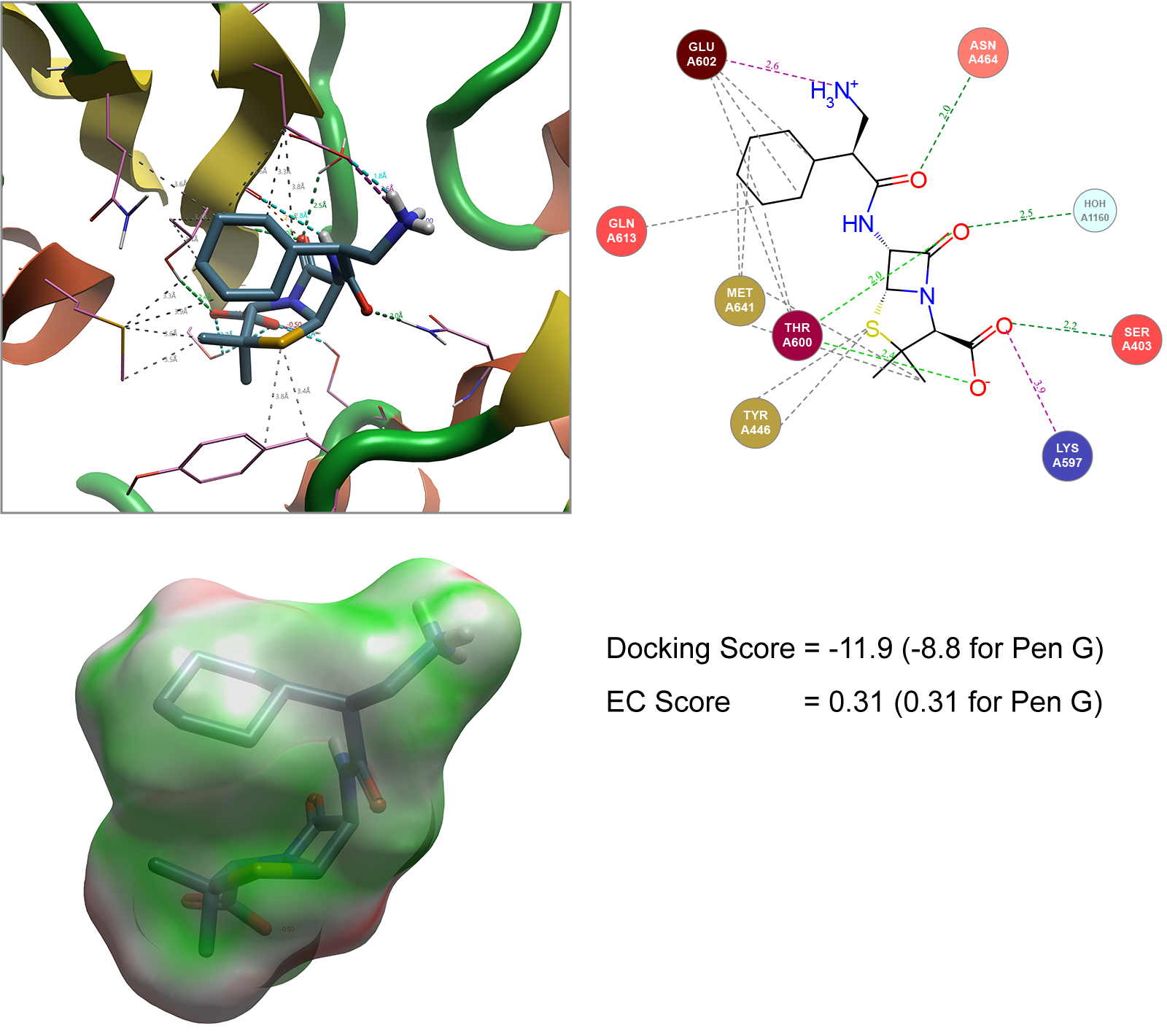
Figure 4. Spark generated ligand bound to Staphylococcus aureus (1MWT). Hydrophobic interactions not shown in Interaction Map (top right) for image clarity. EC image (bottom left) shows areas of good electrostatic complementarity colored in green with areas of poor electrostatic complementarity colored in red. Images generated by Flare.
In ranking these molecules, the saturated example shown in Figure 4 was highlighted as a particularly interesting example. Not only does this ligand show that replacement of the aryl ring with a cyclohexane ring is tolerated but coupled with a 1 carbon chain extension of the amino group compared to ampicillin improves the overall docking score (when compared to penicillin G) while maintaining the electrostatic complementarity of this ligand with the protein.
Engage Cresset Discovery to advance your project
Cresset Discovery have efficiently navigated multiple antibacterial projects in partnership with various clients, underpinning our experience in this space. Our team has extensive virtual screening capabilities. We provide you with comprehensive sampling of available chemical space and, using expert insight and experience in medicinal chemistry to interpret and elucidate the results, deliver an enriched final list of candidate compounds to prioritize for wet screening. Our expert modelers can optimize the starting screening template for better results and can create bespoke compound collections, for example to include natural product databases, to suit your specific project requirements.
Contact Cresset Discovery for a confidential discussion to find out how our intelligent computational approaches can support your research.



















