Identifying novel inhibitors against tuberculosis

Scott Midgley†, Nathan Kidley†
†Cresset, New Cambridge House, Bassingbourn Road, Litlington, Cambridgeshire, SG8 0SS, UK
Abstract
Fragment-based drug discovery is an evolving area of research with unique challenges in developing the hits into lead molecules. In this case study we use the bioisostere solution, Spark™,1 to demonstrate how structure-based virtual screening effectively identifies novel InhA reductase inhibitors for tuberculosis (TB) therapies. We demonstrate how the docking score capabilities in Spark are used to grow a fragment into a new region of the active site to identify new areas of chemistry.
Introduction
TB remains a leading cause of mortality in the 21st century. Enoyl-[acyl-carrier-protein] reductase (InhA) is a popular target in TB small molecule therapy research, however, many of the leading therapeutic compounds require activation via NAD pathways before they can inhibit this enzyme. Blocking this activation is a known TB resistance mechanism, therefore therapies that inhibit InhA without metabolic activation pose an interesting and active area of research. This was the aim of Sabbah et al.4 who performed a fragment-based screening investigation and then optimized the hit fragments. In the present study, we show that Spark can independently identify the experimental hit compounds using the docking method to grow fragments into the InhA active site.
The traditional method for scoring results in Spark is primarily a ligand-based method. Cresset's field similarity technology is used with a shape similarity metric to compare the results to a starter molecule, to identify bioisosteres. Alternatively, Spark can use the Lead Finder™2 docking engine in an alternative scoring process, which calculates a docking score directly from the protein and ligand. This approach enables users to grow ligands into unexplored regions of the active site and identify results making novel interactions with the target protein.
In this case study, we grew the starter ligand taken from PDB: 6SQ5 bound to ‘Site I’ towards ‘Site III’ in the active site (Figure 1), replacing the carboxylic acid moiety whilst preserving its interactions with the protein structure. This was due to limited scope to grow the ligand into ‘Site II’, but growing into ‘Site III’ offered the potential to increase the intrinsic potency of the ligand.
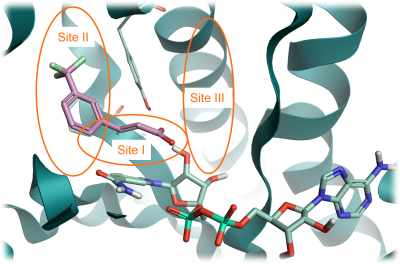
Figure 1. InhA protein from PDB: 6SQ5 with three key sites identified. Site I: catalytic site; Site: II hyrophobic site; Site III: solvent-exposed region. Pink: fragment structure to grow. Pale green: NAD cofactor. Green ribbon: InhA protein.
Computational fragment screening in Spark
Prior to the Spark docking experiments, a traditional Spark field and shape search was performed to identify potential bioisosteres of the carboxilate group of the fragment in Figure 1. This focused the chemical search space in preparation for the Spark docking experiment.
The results from this search identified a range of bioisosteres such as sulfoxide, sulfone, sulfonamide, sulfoximine, imidazole, pyrazole and triazole fragments amongst others. We focussed on the sulfone and sulfonamide bioisosteres of the carboxilate group as they provide a convenient, synthetically accessible handle to synthesize analogues. These fragments also provided a suitable vector to grow the ligands into either Site II or III.
A stepwise approach was then followed for the docking experiments, consisting of three distinct phases.
- Bioisosteric replacement of the carboxylate group. Here, the docking method was used to grow the starter molecule, replacing the carboxylate in the X-ray ligand. The fragment space was filtered to include an SO2 substructure. Furthermore, an automatic fragment size constraint was applied to limit the size of the replacement fragments, to be similar to that of the carboxylate group
- Fragment screening experiment starting from the top-ranking result from phase 1. The Spark docking experiment used the top-ranking sulfone result from phase one as the starter molecule for further fragment growth. The experiment did not use a substructure filter on the fragments but did use the automatic fragment size constraints
- Fragment screening experiment on the top-ranking sulfone result from phase one, using a protein interaction constraint. The docking method was used with a docking constraint, to ensure that the results must contain a H-bond to a key oxygen atom in the NAD molecule in the active site.
Phase one: Bioisosteric replacement of carboxylate group
We used the docking method to identify small bioisosteric replacements of the carboxylate with the intention of growing the ligand in the active site. The docking grid was created from the experimental ligand and the size was set to 8 Å, which is sufficiently large to enable fragments to grow into Site III. The fragment databases searched were the ChEMBL and Commercial Very Common and Common databases which provide nearly 100,000 fragments to search.5,6
A substructure filter requiring an SO2 group to be present in the searched fragments was added as an advanced search option, together with an automatic fragment size constraint to limit the size of the replacement fragments to be similar to that of the carboxylic acid group. This focused the search space to relatively small SO2-containing fragments.
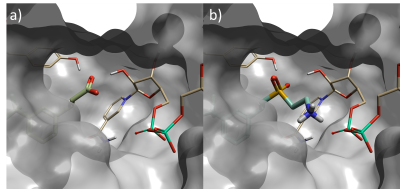
Figure 2. a) Starter molecule (crystallographic ligand) in protein from PDB: 6SQ5, b) Top scoring sulfone compound identified in phase one Spark experiment – fragment is in closer contact with NAD phosphorus atom than starter molecule.
Top scoring results of this first phase yielded multiple hits with an SO2 group directly replacing the original carboxylate. There were additional results where the SO2 was occupying different parts of the active site, or an atom further out from the carboxylate carbon. These variations occur because the SO2 filter is not spatially constrained; this could have been improved by an attachment point constraint.
The top scoring result is shown in Figure 2. The replacement fragment has one of the SO2 oxygen atoms oriented to form a hydrogen-bond with a tyrosine residue in the active site; and a second hydrogen-bond to a hydroxyl group on the NAD sugar, mimicking the interactions made by the carboxylate in the starter molecule. Furthermore, the ethylamine group extends from the sulfone to form an additional hydrogen-bond with phosphate of the NAD cofactor. This is a new interaction which could not have been discovered with the traditional ligand-based method, as the starter ligand did not have atoms in this area of the active site.
Phase two: Second fragment screening experiment on the identified sulfone
The top scoring sulfone-ethylamine compound identified in phase one was used as the starter molecule in this second experiment (Figure 3). The ethylamine group was selected as the fragment to be replaced (blue halo in Figure 3), with the attachment point atom (red circle in Figure 3) constrained to be a nitrogen atom, to ensure that results of this experiment contain only sulfonamide bioisosteres of the original carboxylate moiety. An automatic fragment size constraint were used as an advanced option for this experiment.
.png)
Figure 3. The sulfone-ethylamine compound is the top scoring result from the phase one Spark experiment and was used as an updated starter molecule for phase two. Blue halo: fragment to be replaced. Red circle: attachment point.
As in phase one, the Very Common and Common ChEMBL and Commercial fragment databases were searched, in this case with a 5 Å grid around the new starter molecule. The lower grid size was selected to reduce the computational cost of the docking experiment, while the updated reference molecule facilitates the access to Site III (Figure 1) during the experiment.
Several interesting sulfonamide replacements were identified in phase two (Table 1), including the imidazole and morpholine structures shown in Figures 4a and 4b, forming interactions with the protein and NAD (carbon atoms colored in tan) which are located at a significant distance from the reference structure.
Table 1. Spark Results Table showing top six fragments identified in phase two.
.png)
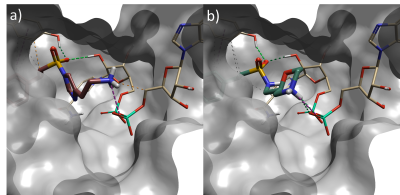
Figure 4. a) Spark result with imidazole sulphonamide (result 4), b) Spark result with morpholine sulphonamide (result 6). Tan: NAD cofactor. Grey surface: Protein active site surface.
Phase three: Third fragment screening experiment on the identified sulfone, applying a protein contact constraint
The Spark experiments ran in phases one and two identified numerous interesting designs. To complement these results, an experiment searching one of the Spark reagent databases was performed to rapidly identify synthetically tractable compounds from common reactions. Sulfonamides were formed by searching the primary aromatic amine fragments (retaining the amine moiety), which provides approximately 24,000 fragments to search6. No filters were applied in this experiment. The default 5 Å docking calculation grid was used, and a docking constraint was added to an NAD oxygen atom, to prioritize compounds that could form hydrogen bonds with the cofactor.
The results from this experiment identified further interesting sulfonamide compounds (Table 2) which can make hydrogen bonds with the constrained NAD oxygen and other protein residues.
Interestingly, the compound with the sixth best docking score carries the same fragment that was identified as the optimal fragment for this pocket by Sabbah et al. As shown in Figure 5, the Spark results for this compound show that the terminal amine forms a hydrogen bond with the NAD oxygen atom that was constrained during docking, and with a nearby backbone carbonyl.
Table 2. Spark Results Table showing top six fragments identified in phase three.
.png)
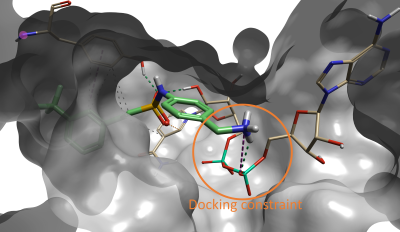
Figure 5. Compound #6 (out of 500) in Spark is the lead compound identified in the paper by Sabbah et al4.
Conclusions
In this case study we used the docking method in Spark to grow the starter ligand from PDB: 6SQ5 into an unoccupied regions of the InhA active site, identifying design ideas with novel ligand-protein binding properties that agree with experimental results published for the InhA TB target.
The docking method extends the capabilities of Spark, enabling to run fragment growing experiments also in those cases where no prior ligand information is available about the desired interactions with the active site.
The use of filters or constraints greatly aids focusing the Spark results to the desired physico-chemical and reaction space. In this case study, filters were applied to prioritize ease of chemical synthesis and increased intrinsic potency via forming new interactions in the active site.
The workflow used for this case study only focused on one area of the active site and two related chemical subseries to generate leads. It could be further expanded by searching alternative reagents, or starting from alternative bioisosteres of the carboxylic acid of the initial fragment hit. Additionally, compounds targeting other regions of the active site could be explored, computationally probing of the binding site to identify alternative chemical series to expand the fragment hit for further development, as would be the approach in a live drug discovery program.
References
- Spark™ V10.7.; Cheeseright T., Mackey M., Rose S., Vinter, A.; Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation, J. Chem. Inf. Model. 2006, 46 (2), 665-676,
- Lead Finder™ (accessed 12 May 2022); BioMolTech®, Toronto, Ontario, Canada
- Cresset science overview (accessed 12 May 2022)
- M. Sabbah, V. Mendes, R. G. Vistal, D. M. G. Dias, M. Záhorszká, K. Mikušová, J. Korduláková, A. G. Coyne, T. L. Blundell and C. Abell; Fragment-Based Design of Mycobacterium tuberculosis InhA Inhibitors, J. Med. Chem., 2020, 63, 4749–4761
- ChEMBL Database (accessed 12 May 2022)
- Products: Screening Compounds - eMolecules, (accessed 12 May 2022)






















