KNIME nodes and Pipeline Pilot components V2.6 released
Version V2.6 of Cresset workflow components for the KNIME™ and Pipeline Pilot™ environments are now available for all desktop solutions.
Spark™ components have been significantly enhanced with the inclusion of the new ‘docking’ feature. This enables Spark to find fragments which pick ligand-protein interactions directly from the protein active site. New example protocols are also available to illustrate the new functionality.
Enhanced functionality for Spark
The new ‘docking’ feature in Spark is available as an add-on module to traditional bioisostere replacement methods. With this method, you can use Spark to grow ligands and fragments into unoccupied pockets of the target protein. You can also use this feature to find result molecules making interactions with the active site of a protein not mapped by an existing starter or reference compound.
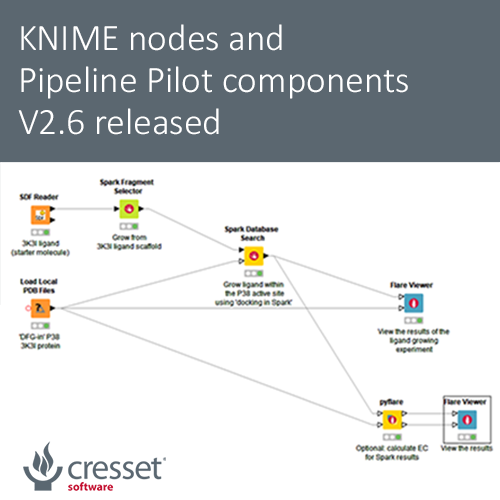
This enhanced functionality was used in the KNIME workflow (Figure 1) based on a P38-alpha case study. In this ligand growing experiment, the docking feature in Spark was used to grow the ligand in PDB:3K3I within the active site of P38-alpha. The objective was to identify potential novel selective and non-ATP competitive inhibitors for P38-alpha.
.png)
Figure 1. Ligand growing workflow using the new docking feature in Spark.
The ligand in PDB:3K3I (Figure 2, left) binds to the inactive, ‘DFG-out’, conformation of P38-alpha. In this form, the activation loop is distorted and the catalytic residues are displaced from their usual position and are thus incapable of binding ATP. This attractive type of non-competitive inhibition is, however, often associated with lack of selectivity towards other kinases which can also adopt this conformation.
In contrast, more selective but ATP-competitive inhibitors like the ligand in PDB:3ROC (Figure 2, right) interact with an active ‘DFG-in’ conformation of P38-alpha. These ligands are more selective as they can make H-bond interactions with Met109 and Gly110, thanks to the plasticity of the hinge region allowing a 180° flip of the peptide bond of Gly110 (Figure 2).
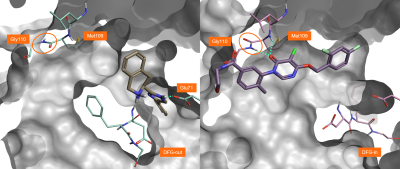
Figure 2. Left: The ligand in PDB:3K3I binds to the inactive form of P38-alpha (‘DFG-out’). Right: The ligand in PDB:3ROC binds to the active form of P38-alpha (‘DFG-in’), making H-bond interactions with Met109 and Gly110.
The PDB:3K3I structure was modeled in Flare™, flipping the peptide bond of Gly110 to mimic the conformation of this residue in PDB:3ROC. The ligand growing experiment was then started using the 3K3I ligand (Figure 3) as the molecule to grow within the modeled 3K3I protein structure, using the docking feature in Spark to score the potential result molecules.
As the experiment starts from the inactive conformation of the protein, Spark result molecules should be non-ATP competitive. At the same time, if they can make H-bond interactions with Gly110 and Met109, they should gain selectivity towards other kinases.
.png)
Figure 3. Ligand in PDB:3K3I is the starter molecule for this ligand growing experiment.
In the ‘Spark database Search’ KNIME node the score method was set to Docking (Figure 4). This activates a set of related options which enable the user to define the region of the active site where to dock the ligands, specify optional docking constraints on H-bond forming atoms on the protein, and define the maximum tolerated penalty for violating a docking constraint.
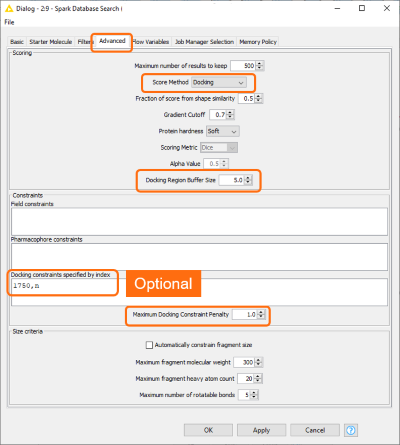
Figure 4. New advanced options in the Spark Database Search KNIME node to activate the ‘docking’ feature.
The search was run on a Spark reagent database of 2K thiols derived from eMolecules building blocks.1 As an optional step (Figure 1, bottom), Spark results can be further refined by calculating their Electrostatic Complementarity™ scores towards the modeled 3K3I protein structure.
The 6 top scoring results from the Spark experiment are shown in Figure 5. All top scoring results can make a good network of aromatic-aromatic and H-bond interactions with P38-alpha, and in particular with Gly110 and Met109. They also show good Electrostatic Complementarity towards the protein, as expressed by their ECr score.
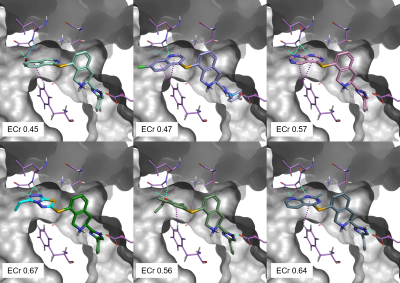
Figure 5. Top scoring results from the Spark ligand growing experiment. Green: H-bond interactions. Cyan: weak H-bond interactions. Purple: aromatic-aromatic interactions.
Of particular interest is result #4 (Figure 5, bottom-left; Figure 6, left), whose thiazolo-triazole moiety makes a similar network of H-bond interactions as the Pfizer compound PF-03715455 in its bioactive conformation from PDB:2YIS (Figure 6, right).
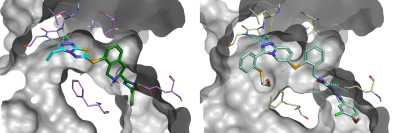
Figure 6. Left: #4 result from the Spark ligand growing experiment. Right: PF-03715455 from PDB:2YIS. Green: H-bond interactions. Cyan: weak H-bond interactions.
New example workflows for KNIME and Pipeline Pilot
Example workflows using the new docking feature in Spark are available for both KNIME and Pipeline Pilot (Figure 7).
Figure 7. Example Pipeline Pilot protocol using the new ‘docking’ feature in Spark.
Other enhancements
Additional enhancements for the Spark components include the option to specify field and pharmacophoric constraints for the reference molecules in traditional Spark experiments based on ligand similarity, and improved fragment selection, now supporting water replacement experiments.
Try the Cresset KNIME and Pipeline Pilot components on your project
Customers can contact support to get these new components free of charge. If you’re not currently using Cresset desktop solutions you can request an evaluation to try them on your project.



















