Cresset: Your partner for virtual screening whether outsourced and/or in-house

High-throughput virtual screening is a powerful computational technique for exploring new chemical space, discovering diverse chemotypes and generating new IP in the search for novel hits. It is far more cost effective than the wet lab alternative, both in terms of ‘per compound’ screening costs (even when running high accuracy docking or equivalent calculations, there are at least 2 orders of magnitude difference in costs from $’s per compound for wet screening to cents per compound for in silico) and likely hit rates (less than 1% for typical HTS whilst in silico methods can be as high as 10%, but on average will be at least 1-2%). Even so, when embarking on an in silico screening project you need to determine the optimum, most efficient approach in terms of time and resources.
When deploying a virtual screening approach, there is a fundamental choice to be made between different methods, or a combination of technologies. The process of searching for novel hits can be broadly divided into two disciplines: structure-based and ligand-based, the choice between them (or the choice to use a combination of the two approaches) can be complex and multi-factorial. We will analyze some of this complexity in the next section.
Ligand-based vs structure-based virtual screening
When we talk about structure-based virtual screening these days, we mostly mean docking: in which we ‘dock’ or attempt to bind ligands against a biological target to prioritize them based on their predicted binding affinities. In order to run a docking experiment, you must have a known 3D structure of the protein target, either obtained by X-ray or NMR analysis, or by using modeling techniques such as homology modeling to predict the likely protein structure and binding mechanism.
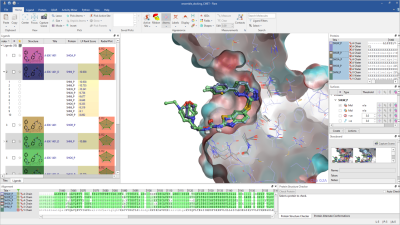
Figure 1: Docking is a structure-based virtual screening method that prioritizes ligands based on their predicted binding affinities to the biological target.
Whilst docking methods require a target structure to proceed, ligand-based virtual screening focuses on the other part of the binding equation: using ligand information to assess or score the likelihood of a compound being a hit when compared to a known active against the target. In addition to being able to be applied in all cases, whether a target protein structure is available or not, ligand-based virtual screening also tends to be a less computationally intensive method. This makes it the preferred option when maximum diversity is required, as it is possible to screen massive chemical spaces in tractable time and at reasonable cost as compared to structure-based methods. There are also cases where ligand-based methods are not preferred, even when there is a known active, for example, if the known active isn’t potent enough, or if it doesn’t exploit all the interactions available in the active site. In these cases, structure-based methods may be preferred.
A combination of ligand-based and structure-based approaches can be useful when designing a virtual screening strategy to exploit all the target information available. This was demonstrated in our recent case study, Finding new leads from peptides and natural ligands, which investigated ligands that could be used as appropriate FOXO-peptide mimetics. Neither technique guarantees to give absolute binding energies or ligand potencies, but they do give good rank ordering of compounds, so are often used as a means of selection and prioritization of ligands for a wet screen. Combining the results of both approaches provides further diversity, returning different hits to give an enriched final list.
How best to progress your virtual screening project
The optimal way for you to progress your virtual screening project will depend on the expertise and resources available to you. There is a strong business case for outsourcing virtual screening as it can bring:
- The scientific knowledge, experience and sophisticated methods required to set up and run experiments, and interpret the results to get maximum output, without the expense of hiring and training an in-house team that may not be required full-time over a period of months or years.
- The computing infrastructure needed to run and manage a high volume of experiments in parallel, allowing you to easily scale up operations and advance your project.
Whilst an attractive option, outsourcing is not always the favored approach, and alternative strategies can work well and still deliver excellent results, such as running virtual screens in-house.
Option 1: Engage Cresset Discovery Services
Cresset Discovery Services CADD scientists can step in to collaborate with your team. We apply our advanced ligand and structure-based solutions to:
- Determine the binding conformation of a ligand needed for a ligand virtual screen
- Verify the assignment and conformation of a ligand within a solved crystal structure
- Use proprietary Cresset technology to provide accurate electrostatic descriptions of your ligand and target
- Select the optimal criteria for the screen through access to a variety of commercial databases
- Filter criteria for the search and restricting the search to certain heavy atom sets
Once the search is complete, you will receive a full analysis of your results, prioritizing and ranking ligand hits as well as the raw data in an appropriate format, for you to analyze using your in-house tools. Our team will then meet with you to present, review, and discuss the analysis, so that you are confident in the meaning of your results and the next steps to take in your project.
Cresset Discovery Services has delivered hundreds of successful virtual screening projects for pharmaceutical, biotechnology, agrochemical, flavor and fragrance companies. Confidentiality does not allow us to reveal all this work, but you can read about a few projects on our website and refer to external sources1.
If you’d like to engage Cresset Discovery Services to advance your virtual screening project, contact us for a free confidential discussion.
Option 2: In-house using Cresset software solutions
If you do already have the required expertise to set up and run virtual screening experiments in-house, and to analyze the results in order to gain value and insight from your experiments, your team can license the latest Cresset software solutions.
Ligand-based virtual screening
Blaze™ is Cresset’s advanced ligand-based virtual screening platform. It returns diverse hits from large chemical databases and has a search algorithm that increases search speed fourfold.
Blaze is designed to be a distributed computing application running on a GPU or CPU based Linux cluster, however, users can access all commands through a web-based graphical user interface or through a RESTful web service API. Blaze virtual screens can also be accessed through workflow tools such as KNIME or Pipeline pilot.
Request an evaluation of Blaze.
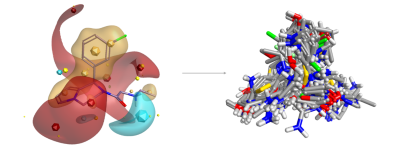
Figure 2: Blaze search using a known A2C adrenergic agonist as query retrieves a wide diversity of novel structures including many known actives.
Structure-based virtual screening
Lead Finder™ implements standard genetic algorithm-based pose generation and a set of proprietary scoring functions. It has been integrated into Flare™, our agile ligand and structure-based design platform, to give a high-throughput docking tool. The algorithm is designed to rapidly dock and rank ligands according to their binding potencies and to separate actives from inactives.
In Flare, users can access the docking commands through the desktop GUI. All Flare functionality, including docking with Lead Finder, can be accessed through the Python™ API, which has the added benefit of allowing users to automate tasks and customize the interface to their needs. Lead Finder can also be accessed through KNIME and Pipeline Pilot workflow tools.
Request an evaluation of Lead Finder.
Users of all Cresset solutions receive training and have access to support for installation and application. However, maintaining the hardware and software required to run large-scale virtual screens requires technical capabilities and internal infrastructure. We recognize that sometimes these physical resources are either not available or are not to a sufficient scale for the experiments required to be run. In these cases, there is a mixed model where customers can benefit from Cresset’s extensive HPC infrastructure, as we explain below:
Option 3: Run virtual screens using a Cresset managed cluster or cloud infrastructure
Cresset can offer technical support by providing access to high performance computing (HPC) resources. This could be through a computer cluster, or a managed cloud infrastructure such as Amazon Web Services (AWS). In this approach, software and databases are maintained by Cresset and there is no need for in-house IT support, but it does not eliminate the need for in-house computational chemistry expertise to process and evaluate virtual screen results.
The Blaze Cloud implementation provides access to a secure Cresset hosted portal which provides access to a single-tenant Blaze environment configured to your requirements. There is also an Amazon cloud-ready version for deployment. Users access the Blaze Cloud virtual screening platform through the Cresset cluster, either through the webservice API, or the web application interface. The screening database available through the Cresset Blaze server contains more than 18 million compounds. These are molecules from commonly available vendor databases, such as Asinex, Enamine, Maybridge, Otava, Molport, etc. which can be readily ordered.
Sign up for access to Blaze Demo server.
Conclusion
Whether you already have a dedicated team and resources, or you are new to virtual screening, Cresset can work with you to devise the best approach for your project. If you’re still unsure as to the best approach for your project, contact us for a free confidential discussion.
References1
- Structure-Based Library Design and Fragment Screening for the Identification of Reversible Complement Factor D Protease Inhibitors, Vulpetti, A. et al. J. Med. Chem., 60 1946, 2017
- Discovery of Compounds that Positively Modulate the High Affinity Choline Transporter, Choudhary et al. Frontiers in Molecular Neuroscience 2017 Feb 27; 10:40
- Tioxazafen: A New Broad-Spectrum Seed Treatment Nematicide, Slomczynska, U. et al. Discovery and Synthesis of Crop Protection Products, 2015
- Selectivity of Kinase Inhibitor Fragments, Bamborough et al. J. Med. Chem. 54, 5131, 2011
- Modulation of 11β-Hydroxysteroid Dehydrogenase Type 1 Activity by 1,5-Substituted 1HTetrazoles, Webster, S.P. et al. BMCL 20(11) 3265-3271, 2010
- Novel Lead Structures for p38 MAP Kinase via Field Screen Virtual Screening, Cheeseright, T. C. et al. J. Med. Chem., 52, 4200-4209, 2009



















